|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Induction of general anesthesia reduces FRC. Loss of tonic parasternal intercostal muscle activity,[272] [273] [274] development of phasic expiratory activity of respiratory muscles ( Fig. 6-16 ), [274] alteration in diaphragm position,[273] and changes in thoracic blood volume have been proposed as mechanisms responsible for anesthetic-induced decreases in FRC.[274] There is a relative sparing of diaphragmatic activity compared with intercostal muscle function during halothane anesthesia.[272] [273] A change in diaphragmatic shape occurs during anesthesia, with dependent regions shifting cephalad and nondependent regions shifting caudad ( Fig. 6-17 ).[275] [276] [277] Anesthesia-enhanced expiratory muscle activity results in inward displacement of the rib cage, an action that contributes to the reduction in FRC. In contrast, inspiratory rib cage expansion may remain relatively well preserved because of phasic inspiratory activity of scalene muscles despite the attenuation of parasternal intercostal muscle activity.[273] Areas of atelectasis in dependent lung regions frequently occur in anesthetized, spontaneously ventilating humans, and they may be related to alterations in respiratory muscle tone but not specifically to any single chest wall structure.[275] There is no direct correlation between changes in FRC and anesthesia-induced atelectasis. [277] Atelectasis markedly contributes to gas exchange abnormalities by increasing intrapulmonary shunting and may be reduced by the application of positive end-expiratory pressure or phrenic nerve stimulation of the diaphragm. [278]
The reasons for the differential effects of inhaled anesthetics on inspiratory and expiratory respiratory muscles
 Figure 6-16
Representative record from one subject while awake and
during halothane anesthesia. The three upper tracings
are electromyograms. Lower tracings represent ribcage
and abdominal dimensions measured by respiratory impedance plethysmography. Open
and solid circles denote the beginning and end of inspiration, respectively.
Notice that the amplitudes of ribcage and abdominal excursions diminish during halothane
but that the relationship between their amplitudes is preserved. (From Warner
DO, Warner MA, Ritman EL: Human chest wall function while awake and during halothane
anesthesia. I. Quiet breathing. Anesthesiology 82:6, 1995.)
Figure 6-16
Representative record from one subject while awake and
during halothane anesthesia. The three upper tracings
are electromyograms. Lower tracings represent ribcage
and abdominal dimensions measured by respiratory impedance plethysmography. Open
and solid circles denote the beginning and end of inspiration, respectively.
Notice that the amplitudes of ribcage and abdominal excursions diminish during halothane
but that the relationship between their amplitudes is preserved. (From Warner
DO, Warner MA, Ritman EL: Human chest wall function while awake and during halothane
anesthesia. I. Quiet breathing. Anesthesiology 82:6, 1995.)
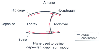 Figure 6-17
Diagram of a midsagittal section of the thorax while
awake (solid lines) and while anesthetized (red
lines) with a 1.2 minimum alveolar concentration (MAC) of halothane.
Chest wall configuration was determined using images of the thorax obtained by three-dimensional
fast computed tomography. (Adapted from Warner DO, Warner MA, Ritman EL:
Atelectasis and chest wall shape during halothane anesthesia. Anesthesiology 85:49,
1996.)
Figure 6-17
Diagram of a midsagittal section of the thorax while
awake (solid lines) and while anesthetized (red
lines) with a 1.2 minimum alveolar concentration (MAC) of halothane.
Chest wall configuration was determined using images of the thorax obtained by three-dimensional
fast computed tomography. (Adapted from Warner DO, Warner MA, Ritman EL:
Atelectasis and chest wall shape during halothane anesthesia. Anesthesiology 85:49,
1996.)
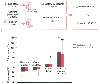 Figure 6-18
A, Schematic diagram of
the effects of volatile anesthetics on synaptic transmission to expiratory premotor
neurons in the caudal ventral respiratory group. The overall glutaminergic excitatory
drive is reduced without effecting NMDA receptor function, and overall inhibition
is increased because of an increase in GABAA
receptor function. Presynaptic
inhibitory drive is also reduced, leading to a decrease in control discharge frequency
of the premotor neuron. Red arrows represent anesthetic
effects. B, Effects of halothane and sevoflurane
on excitatory and inhibitory neurotransmission to canine medullary expiratory neurons.
Mean changes ± SD are given for neuronal control frequency, overall excitation,
overall inhibition and inhibitory receptor response by 1 minimum alveolar concentration
(MAC) of halothane and sevoflurane (*P < .05,
** P < .01, *** P
< .001 versus no change). (Adapted from Stucke AG, Stuth EAE, Tonkovic-Capin
V, et al: Effects of halothane and sevoflurane on inhibitory neurotransmission to
medullary expiratory neurons in a decerebrate dog model. Anesthesiology 96:955,
2002.)
Figure 6-18
A, Schematic diagram of
the effects of volatile anesthetics on synaptic transmission to expiratory premotor
neurons in the caudal ventral respiratory group. The overall glutaminergic excitatory
drive is reduced without effecting NMDA receptor function, and overall inhibition
is increased because of an increase in GABAA
receptor function. Presynaptic
inhibitory drive is also reduced, leading to a decrease in control discharge frequency
of the premotor neuron. Red arrows represent anesthetic
effects. B, Effects of halothane and sevoflurane
on excitatory and inhibitory neurotransmission to canine medullary expiratory neurons.
Mean changes ± SD are given for neuronal control frequency, overall excitation,
overall inhibition and inhibitory receptor response by 1 minimum alveolar concentration
(MAC) of halothane and sevoflurane (*P < .05,
** P < .01, *** P
< .001 versus no change). (Adapted from Stucke AG, Stuth EAE, Tonkovic-Capin
V, et al: Effects of halothane and sevoflurane on inhibitory neurotransmission to
medullary expiratory neurons in a decerebrate dog model. Anesthesiology 96:955,
2002.)
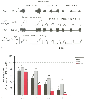 Figure 6-19
Effect of increasing halothane on phrenic nerve and neuron
activity. With increasing anesthetic depth, both peak phrenic nerve (PPA) and peak
neuron activities are depressed. PPA shows the greatest sensitivity to halothane.
A, Phrenic nerve activity and the discharge frequency
of an inspiratory bulbospinal neuron. Inspiratory (phrenic burst) duration decreases
with increasing anesthetic depth. B, Peak expiratory
neuron (E), peak inspiratory neuron (I) activity, and PPA. Expiratory neuronal activity
was most resistant to the effects of halothane. Bars
show the normalized mean ± SEM, and the numbers above the bars indicate the
numbers of neurons studied for each condition. (Adapted from Stuth EAE,
Tonkovic-Capin M, Kampine JP, et al: Dose-dependent effects of halothane on expiratory
and inspiratory bulbospinal neurons and the phrenic nerve activities in dogs. Anesthesiology
81:1470, 1994.)
Figure 6-19
Effect of increasing halothane on phrenic nerve and neuron
activity. With increasing anesthetic depth, both peak phrenic nerve (PPA) and peak
neuron activities are depressed. PPA shows the greatest sensitivity to halothane.
A, Phrenic nerve activity and the discharge frequency
of an inspiratory bulbospinal neuron. Inspiratory (phrenic burst) duration decreases
with increasing anesthetic depth. B, Peak expiratory
neuron (E), peak inspiratory neuron (I) activity, and PPA. Expiratory neuronal activity
was most resistant to the effects of halothane. Bars
show the normalized mean ± SEM, and the numbers above the bars indicate the
numbers of neurons studied for each condition. (Adapted from Stuth EAE,
Tonkovic-Capin M, Kampine JP, et al: Dose-dependent effects of halothane on expiratory
and inspiratory bulbospinal neurons and the phrenic nerve activities in dogs. Anesthesiology
81:1470, 1994.)
Volatile anesthetics also differentially depress neuromuscular transmission and skeletal muscle contractility. Halothane impairs excitation-contraction coupling and neuromuscular transmission to equivalent degrees, whereas isoflurane, sevoflurane, and enflurane depress diaphragmatic contraction primarily through alterations in neuromuscular transmission.[284] [285] [286] Isoflurane, enflurane, and sevoflurane depress the tension of the diaphragm in response to phrenic nerve stimulation.[284] [287] Although these results may partially explain clinical observations in humans, a wide range of species variations do exist. For example, volatile anesthetics decrease expiratory muscle activity and maintain parasternal intercostal activity in dogs, but converse actions are observed in humans.[273] [274] [275] Similar to volatile agents, nitrous oxide affects chest wall function and breathing by changing the distribution and timing of neural drive to the respiratory muscles.[288] Nitrous oxide decreases tidal volume as a result of a reduction in rib cage motion and increased phasic expiratory activity in humans. In contrast, xenon does not affect transdiaphragmatic pressure or diaphragmatic electromyography.[289]
Changes in chest wall positions produce alterations in efferent impulses from stretch receptors in intercostal muscle spindles that act to maintain relatively constant tidal volume during variations in inspiratory resistance. Increases in spindle discharge enhance motor activity to muscle fibers until muscle shortening relieves tension in the spindles. With increased inspiratory resistance, the muscle spindles detect a failure of shortening by the appropriate amount, and afferent signals are subsequently increased to the motor neuron pool. Accessory muscles of inspiration may be recruited as well. This reflex increase in inspiratory effort sustains tidal volume and minute ventilation despite a greater inspiratory resistive load. These and other forces maintain normal ventilation with changes in body position, inspiratory resistance, and compliance. The extrapolation of these data obtained in experimental animals to humans should be approached with caution. For example, tension receptors in the diaphragm, not the muscle spindles, induce reflex inhibition of inspiratory intercostal activity associated with a decrease in inspiratory duration in dogs but not in humans. This effect appears to be mediated at the supraspinal level.[290]
Ventilation is maintained during halothane anesthesia despite increases in resistance produced by partial endotracheal tube occlusion or an acute reduction in lung compliance until a threshold level is reached.[291] Subsequently, minute ventilation falls, and retention of
Expiration is passively affected by the recoil characteristics of the lung during normal breathing. In anesthetized patients, the ventilatory response to expiratory resistance is reduced to a greater extent than is the response to inspiratory resistance. Conscious and anesthetized humans exhibit decreases in respiratory rate during expiratory resistive loads, but only anesthetized subjects develop rib cage-abdominal wall motion asynchrony resulting in less effective ventilation and increases in carbon dioxide.[296] This concept may be particularly important in spontaneously breathing, anesthetized patients who demonstrate expiratory obstruction (e.g., partial breathing circuit occlusion, asthma, emphysema). Patients with COPD who do not retain carbon dioxide in the conscious state display a greater degree of hypoventilation awake than healthy patients during halothane anesthesia, consistent with these observations ( Fig. 6-20 ).[297] Paradoxically, patients with COPD develop less atelectasis and have greater preservation of gas exchange compared with healthy patients, despite similar diaphragmatic displacements during anesthesia. [298] The mechanisms for this observation are unclear, but intrinsic positive end-expiratory pressures, altered patterns of respiratory muscle activation, or altered ventilation-perfusion mismatch may play important roles. [277]
|
|
|
|
|
|
|
|
|
|
|
|
|