|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In the broadest sense, a receptor is a component of a cell that interacts selectively with an extracellular compound to initiate a cascade of biochemical events that culminate in the observed effects of the compound. Binding of a drug to a receptor determines (1) the quantitative relationship between a given dose of a drug and the resulting effect, (2) the selectivity of a given drug's activity and effect, and (3) an explanation of the pharmacologic activity of receptor agonists, antagonists, and inverse agonists. Receptors therefore mediate or amplify the effect of a drug on the biologic system.
Although the overall concept of receptors is generally attributed to Paul Ehrlich (1854–1915), earlier work performed by Claude Bernard (1813–1878) paved the way for receptor theory. This earlier work is of particular interest to anesthesiologists because it elucidated the mechanism of action of curare, an arrow poison. In his experiments, Claude Bernard ligated vessels leading to one hind limb of a frog while leaving nerve input intact; he then administered intravenous curare. Pinching the paralyzed hind limb produced reflex movements in the opposite unparalyzed vessel-ligated hind limb. These experiments and others demonstrated for the first time the separation between sensory and motor nervous systems and revealed that circulating "substances" produced selective effects on organ systems, a concept important in the development of receptor theory.
The concept of selectivity of effect eventually led Paul Ehrlich to his conclusion that "agents cannot act unless they are bound," a cornerstone of receptor theory. Although Ehrlich is attributed with discovering this concept, his contemporary J. N. Langley (1852–1926) coined the term receptive substance (i.e., receptor). Langley extended Claude Bernard's work by showing that nicotine and curare had mutually antagonistic effects on the same receptive substance, which was neither nerve nor muscle. We now recognize this receptive substance as the nicotinic acetylcholine receptor in the neuromuscular junction. Several decades of research have refined the receptor theory, and receptors are now recognized as discrete excitable proteins.
The binding of a ligand (i.e., drug molecule) to its receptor
follows the laws of chemical reactions (analogous to Michaelis Menten enzyme kinetics)
and can be summarized by the following relationship:
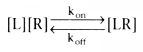
In this reversible equation, kon
is the rate constant for the ligand binding
to the receptor, koff
is the rate constant for the ligand disassociating
from the receptor, [L] is the concentration of the ligand, [R] is the concentration
of the unbound receptor, and [LR] is the concentration of the bound receptor. Units
for kon
are units of [L] multiplied by the units of [R] over time; typically,
units of L are nanomoles per liter. The units for koff
are the units
of [LR] over time. The term Kd
, the dissociation constant, mathematically
defines characteristics of ligand-receptor interactions at equilibrium:
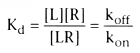
In practice, it is difficult to measure precise drug-receptor occupancy. It is often assumed that the drug-receptor complex represents an intermediate step in the production of a specific effect. Many investigators apply pharmacodynamic theory to compare a given dose of a drug with a resulting effect. An effect can be any biochemical or physiologic variable that is measurable. A measured effect can be an alteration in a biochemical compound, an enzyme level, a physiologic variable such as heart rate or blood pressure, or a response to any graded input into the biologic system. For example, in evaluating the pharmacodynamics of muscle relaxants, the measured effect is not a direct measurement of drug-receptor complexes, but rather a response to a neuromuscular stimulus as delivered by a nerve stimulator.
From the discussion of pharmacokinetics, the delivery of a drug to a given receptor is clearly time and dose dependent. If the receptor is located within the central compartment, there may be nearly instantaneous equilibration after an intravenous injection, and the peak effect may occur immediately. If drug must cross from the central compartment to the site of drug effect, a delay will occur. The onset of drug effect can also be delayed by the time required for the body to respond to the drug. This delay may occur anywhere on the path between drug-receptor binding and clinical manifestation of drug effect. Examples of post-transduction time delays are drug-receptor complex-induced secondary messenger changes, increased enzyme synthesis, and the time required for physiologic change (e.g., reduced fluid content).
Drugs that are agonists induce an effect that mimics endogenous hormones or neurotransmitters when bound to a receptor. This effect may be stimulatory or inhibitory. The term affinity as related to a given agonist is a measure of the attraction between the given drug and receptor. A drug with low affinity for a given receptor tends not to bind to the receptor and produces little or no effect. An agonist that binds avidly (or with high affinity) to a given receptor produces the receptor-determined effect at a lower given dose.
Full agonists completely activate a receptor, whereas partial agonists only partially activate a receptor, even at very high concentrations ( Fig. 3-23 ). The difference between full and partial agonists results from different intrinsic efficacy for each drug. Efficacy should not be confused with affinity. Two drugs can have identical affinity for a receptor (and therefore bind to the same extent at a given
 Figure 3-23
Effects of various types of ligands on receptor responses.
A full agonist produces complete (100%) activation of a receptor at high concentrations,
whereas partial agonist binding results in less than 100% activation, even at very
high concentrations. A neutral antagonist has no activity of its own. Inverse agonists
can be thought of as "superantagonists" because binding of these ligands produces
a response below the baseline response measured in the absence of drug. If the physiologic
effect of the baseline levels of activated receptor (R*) is small, antagonists
and inverse agonists may not be clinically distinguishable.
Figure 3-23
Effects of various types of ligands on receptor responses.
A full agonist produces complete (100%) activation of a receptor at high concentrations,
whereas partial agonist binding results in less than 100% activation, even at very
high concentrations. A neutral antagonist has no activity of its own. Inverse agonists
can be thought of as "superantagonists" because binding of these ligands produces
a response below the baseline response measured in the absence of drug. If the physiologic
effect of the baseline levels of activated receptor (R*) is small, antagonists
and inverse agonists may not be clinically distinguishable.
Drugs that are antagonists inhibit or prevent receptor-mediated agonist effects by competing for receptor occupancy. A competitive antagonist can generally be displaced from the receptor complex by the administration of a large enough concentration of receptor agonist, permitting the agonist to produce the expected effect despite the presence of antagonist. A noncompetitive antagonist usually binds irreversibly to the receptor complex, producing loss of the expected effect that cannot be compensated by administration of high concentrations of receptor agonist. For example, vecuronium is a competitive antagonist of acetylcholine (see Chapter 13 ). Acetylcholine mediates muscle contraction through the postsynaptic nicotinic acetylcholine receptor at the neuromuscular junction. When vecuronium binds to the postsynaptic acetylcholine receptor, acetylcholine agonism is blocked, and neuromuscular transmission is inhibited. The result is flaccid paralysis. Neuromuscular transmission may be reinstated by administering an acetylcholine esterase inhibitor. Acetylcholine esterase inhibitors prevent breakdown of acetylcholine, effectively raising the concentration of the
Inverse agonists can be thought of as "superantagonists" because they decrease receptor responses to less than baseline values (see Fig. 3-23 ). An example of concentration-response relationships for four different benzodiazepines and EEG response[10] is shown in Figure 3-24 (see Chapter 10 ). In this example, the EEG response represents a surrogate clinical end point for binding of benzodiazepines to the γ-aminobutyric acid (GABAA ) ligand-gated ion channel complex with resultant modulations of ion fluxes in the central nervous system (CNS).
Classic receptor theory describes interaction between ligand and receptor based on the laws of mass action. At a molecular level, this interaction has been interpreted over the years to suggest that binding of agonist to receptor initiates a change in receptor conformation, changing the receptor from an inactive (R) to an
 Figure 3-24
The concentration-electroencephalographic (EEG) response
relationship for four benzodiazepines: midazolam (full agonist, a), bretazenil (partial
agonist, b), flumazenil (antagonist, c), and R0 19-4063 (inverse agonist, d). The
maximum effect seen in the EEG response correlates with clinical action (full agonist
> partial agonist > antagonist > inverse agonist). (Adapted from
Shafer S: Principles of pharmacokinetics and pharmacodynamics. In
Longnecker DE, Tinker JH, Morgan GE [eds]: Principles and Practice of Anesthesiology,
2nd ed. St. Louis, Mosby-Year Book, 1997, and from Mandema JW, Kuck MT, Danhof M:
Differences in intrinsic efficacy of benzodiazepines are reflected in their concentration-EEG
effect relationship. Br J Pharmacol 105:164–170, 1992.)
Figure 3-24
The concentration-electroencephalographic (EEG) response
relationship for four benzodiazepines: midazolam (full agonist, a), bretazenil (partial
agonist, b), flumazenil (antagonist, c), and R0 19-4063 (inverse agonist, d). The
maximum effect seen in the EEG response correlates with clinical action (full agonist
> partial agonist > antagonist > inverse agonist). (Adapted from
Shafer S: Principles of pharmacokinetics and pharmacodynamics. In
Longnecker DE, Tinker JH, Morgan GE [eds]: Principles and Practice of Anesthesiology,
2nd ed. St. Louis, Mosby-Year Book, 1997, and from Mandema JW, Kuck MT, Danhof M:
Differences in intrinsic efficacy of benzodiazepines are reflected in their concentration-EEG
effect relationship. Br J Pharmacol 105:164–170, 1992.)
Transgenic animal experiments have shed light on ligand and receptor interactions. In an elegant set of experiments in which β2 -adrenergic receptors were massively overexpressed in mouse myocardium, second messenger responses were increased in the absence of ligand when compared with normal mouse myocardium. Activity of adenylyl cyclase, the second messenger most commonly studied for β2 -adrenergic receptors, is identical at baseline in transgenic animals (in the absence of drug) compared with normal mice stimulated with isoproterenol, a β2 -adrenergic-receptor agonist. Isoproterenol stimulation of transgenic animals is unable to increase adenylyl cyclase activity higher than this elevated baseline. This finding suggests that a subpopulation of receptors is already fully activated in these transgenic animals. One way to analyze these findings is to consider that at baseline a small
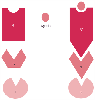 Figure 3-25
Schematic drawing of classic receptor activation. For
years, binding of ligand was thought to cause receptors to change from an inactive
state (R) to an activated state (R*). The activated receptor interacted with
intermediary guanine nucleotide (G) proteins or directly with second messenger cascades
(i.e., effectors [E]), or both. Later information suggests a more complicated equilibrium
between R and R*, as shown in Figure 2-26 and Figure 2-27.
Figure 3-25
Schematic drawing of classic receptor activation. For
years, binding of ligand was thought to cause receptors to change from an inactive
state (R) to an activated state (R*). The activated receptor interacted with
intermediary guanine nucleotide (G) proteins or directly with second messenger cascades
(i.e., effectors [E]), or both. Later information suggests a more complicated equilibrium
between R and R*, as shown in Figure 2-26 and Figure 2-27.
These findings have broad implications for understanding receptor states and drug-receptor interactions. Instead of ligand binding causing a change from R to R* (see Fig. 3-25 ), these data suggest spontaneous conversion from the R to the R* state ( Fig. 3-26 ). In most tissues, the concentration of R* represents a small fraction of total overall receptor (hence the large arrow pointing toward R in Fig. 3-26 ). However, baseline receptor responses are
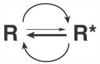 Figure 3-26
Spontaneous conversion of receptors from an inactive
(R) to an active (R*) state. In most tissues, R* represents a small fraction
of the total receptor population.
Figure 3-26
Spontaneous conversion of receptors from an inactive
(R) to an active (R*) state. In most tissues, R* represents a small fraction
of the total receptor population.
The discovery of spontaneous conversion between R and R* has implications for understanding the action of ligands (i.e., agonists, antagonists, and inverse agonists). Instead of agonists causing conversion from R to R*, it is thought that ligands stabilize a given receptor state. As shown in Figure 3-27 , agonists stabilize (or energetically favor) R*, and inverse agonists stabilize R, whereas neutral antagonists bind equally to R and R*. Binding of an agonist to a receptor removes the receptor from the equation because rapid coupling to second messengers occurs, driving (by mass action) the equation toward that specific state ( Fig. 3-28 ). The probability of being in the R* state increases in the presence of an agonist. Conversely, inverse agonists drive the equation to the left, stabilizing the R configuration. Neutral antagonists bind equally to R and R*, preventing agonist binding without altering the equilibrium between R and R*; neutral antagonists do not change the observed baseline response (see Fig. 3-23 ). Many classic antagonists have been tested and found to be inverse agonists rather than neutral antagonists. For example, among the commonly clinically used β2 -adrenergic-receptor (β2 AR) antagonists, metoprolol and bisoprolol show significant inverse agonist activity, carvedilol has weaker inverse agonist properties at the β2 AR and displays
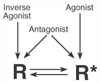 Figure 3-27
Ligand stabilization of receptor states. Agonists stabilize
active receptors (R*), inverse agonists stabilize inactive receptors (R), and
neutral antagonists bind equally to R and R* without affecting the equilibrium
between the two receptor states.
Figure 3-27
Ligand stabilization of receptor states. Agonists stabilize
active receptors (R*), inverse agonists stabilize inactive receptors (R), and
neutral antagonists bind equally to R and R* without affecting the equilibrium
between the two receptor states.
 Figure 3-28
Equilibrium between inactive receptors (R) and active
receptors (R*) is tissue specific and depends on the type of ligand administered.
By stabilizing R*, agonists drive the equilibrium to the right. Inverse agonists
stabilize R, driving the equilibrium to the left. Because neutral antagonists bind
equally to R and R*, they do not affect tissue-specific equilibrium between R
and R*.
Figure 3-28
Equilibrium between inactive receptors (R) and active
receptors (R*) is tissue specific and depends on the type of ligand administered.
By stabilizing R*, agonists drive the equilibrium to the right. Inverse agonists
stabilize R, driving the equilibrium to the left. Because neutral antagonists bind
equally to R and R*, they do not affect tissue-specific equilibrium between R
and R*.
Receptors are located in many places in the cell—outer cell membrane, cytoplasm, intracellular organelle membranes, and nucleus. The overall physical structure of a
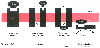 Figure 3-29
Schematic drawing of four types of membrane-associated
drug targets: membrane (G protein-coupled) receptor, ligand-gated ion channel, voltage-sensitive
ion channel, and an enzyme. Potential sites of drug action are shown. βAR,
β-adrenergic receptor; GABAA
, γ-aminobutyric acid.
Figure 3-29
Schematic drawing of four types of membrane-associated
drug targets: membrane (G protein-coupled) receptor, ligand-gated ion channel, voltage-sensitive
ion channel, and an enzyme. Potential sites of drug action are shown. βAR,
β-adrenergic receptor; GABAA
, γ-aminobutyric acid.
One of the most common methods by which membrane receptors translate agonist occupancy into intracellular action is through intermediary G proteins. As a result, G protein-coupled receptors are the most abundant type of receptor known. The overall structure of a G protein-coupled receptor is shown schematically in Figure 3-30 . Two-dimensional representation reveals an extracellular amino terminus containing glycosylation sites, an intracellular carboxyl terminus, a fatty acid attachment (usually
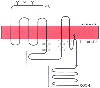 Figure 3-30
Schematic drawing of a G protein-coupled receptor. A
two-dimensional version of receptor structure is shown with seven transmembrane domains,
an extracellular amino (NH2
) terminus (with associated glycosylation sites
[Y]), an intracellular carboxyl (COOH) terminus, palmitoylated cysteine residue (crooked
line extending into the membrane), three extracellular loops, and four
intracellular loops. Major sites of G protein interactions with the receptor are
speckled; potential sites of phosphorylation in the third intracellular loop and
carboxyl terminus are enclosed in boxes.
Figure 3-30
Schematic drawing of a G protein-coupled receptor. A
two-dimensional version of receptor structure is shown with seven transmembrane domains,
an extracellular amino (NH2
) terminus (with associated glycosylation sites
[Y]), an intracellular carboxyl (COOH) terminus, palmitoylated cysteine residue (crooked
line extending into the membrane), three extracellular loops, and four
intracellular loops. Major sites of G protein interactions with the receptor are
speckled; potential sites of phosphorylation in the third intracellular loop and
carboxyl terminus are enclosed in boxes.
Although a two-dimensional schematic is shown in Figure 3-30 , it is important to remember that G protein-coupled receptors are three-dimensional structures with transmembrane segments coalescing around a central binding pocket ( Fig. 3-31 ). [13] Although transmembrane amino acids must be hydrophobic overall to be energetically favored in the lipid membrane, side chains on individual amino acids may be charged. Specific amino acids therefore act as counter ions to anchor charged (water-soluble) hormone or drug to the receptor. Structure-activity relationships between drug and the three-dimensional configuration of the receptor's binding site are critically important in determining binding properties. The chemical structure of a drug must match the three-dimensional configuration of the binding area of a receptor. Subtle changes in drug structure may dramatically alter the ability of a drug to bind to a specific receptor population. Two drugs with seemingly unrelated two-dimensional chemical structures may bind to the same receptor if their three-dimensional structures and charged areas are similar. The three-dimensional components of a ligand that interact with ligand-recognition portions of a receptor are referred to as the pharmacophore.
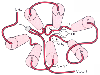 Figure 3-31
Schematic drawing of the three-dimensional structure
of the β2
-adrenergic receptor (a prototypical G protein-coupled receptor),
looking from the outside of the cell inward. Transmembrane domains (denoted by cylinders
and numbered with roman numerals) coalesce to form a binding pocket. The correct
orientation of the agonist norepinephrine is shown. Notice that agonist affinity
is determined by specific amino acids located in transmembranes III, IV, and VII.
These critical sites in transmembrane domains have been determined experimentally
using chimeric receptor and mutagenesis approaches combined with sophisticated computer-modeling
techniques. Ultimate confirmation of these structures will require crystallographic
data, an approach that is being attempted by several laboratories. (Adapted
from Stapelfeldt WH: The autonomic nervous system. In
Schwinn DA [ed]: Scientific Principles of Anesthesia, vol 2. Philadelphia, Current
Medicine, 1998.)
Figure 3-31
Schematic drawing of the three-dimensional structure
of the β2
-adrenergic receptor (a prototypical G protein-coupled receptor),
looking from the outside of the cell inward. Transmembrane domains (denoted by cylinders
and numbered with roman numerals) coalesce to form a binding pocket. The correct
orientation of the agonist norepinephrine is shown. Notice that agonist affinity
is determined by specific amino acids located in transmembranes III, IV, and VII.
These critical sites in transmembrane domains have been determined experimentally
using chimeric receptor and mutagenesis approaches combined with sophisticated computer-modeling
techniques. Ultimate confirmation of these structures will require crystallographic
data, an approach that is being attempted by several laboratories. (Adapted
from Stapelfeldt WH: The autonomic nervous system. In
Schwinn DA [ed]: Scientific Principles of Anesthesia, vol 2. Philadelphia, Current
Medicine, 1998.)
In addition to membrane receptors, pharmacologic agents act on other excitable cell-membrane proteins, such as ion channels. These channels mediate neural signaling by modulating ion permeability in electrically excitable membranes. Because many drugs act directly or indirectly through ion channels, it is important for the anesthesiologist to understand their general function. Classic ion channels such as the sodium channel have charged regions that span the cell membrane. The formation of ion pairs between many positive and negative charges helps to stabilize the channel in the membrane. Voltage-dependent gating of the sodium channel is made possible by the presence of a voltage sensor—a collection of charges that moves under the influence of the cell-membrane electrical field (hence the name voltage-gated ion channels). During depolarization, these charges presumably move outward, causing conformational changes and rearrangement of ion pairs that result in sodium permeability. Local anesthetics work by blocking voltage-gated sodium channels. This concept is discussed in more detail in Chapter 14 .
The combination of a classic receptor protein plus an ion channel results in a ligand-gated ion channel, giving rise to the unique ability of certain drugs to alter membrane permeability to ions directly. Many anesthetic drugs act on ligand-gated ion channels, such as the nicotinic acetylcholine receptor and the GABAA receptor. The structure of the GABAA receptor is shown in Figure 3-32 .[14] [15] Ligand-gated ion channels have hydrophobic and charged
 Figure 3-32
Cross-section of a schematized γ-aminobutyric acid
(GABA)-benzodiazepine receptor complex, which is a prototypical ligand-gated ion
channel complex. Binding of benzodiazepine agonists to the GABAA
ligand-gated
ion channel facilitates the action of endogenous GABA. This results in increased
inhibitory chloride ion flux in the central nervous system. A,
Schematic drawing of the GABAA
ligand-gated ion channel complex shows
the receptor sites for benzodiazepines and GABA, as well as distinct receptor sites
for barbiturates and alcohol. B, Model of the GABAA
receptor and chloride ion channel shows that the protein complex is composed of a
hetero-oligomer of five subunits, including α, β, γ, and δ
or ρ polypeptides. Each subunit has four putative membrane-spanning domains
(numbered 1 through 4 and represented by cylinders). (A,
from Berkowitz DE: Cellular signal transduction. In
Schwinn DA [ed]: Scientific Principles of Anesthesia, vol 2. Philadelphia, Current
Medicine, 1998; B, adapted from Firestone L, Quinlan
J, Gyulai F: Mechanisms of anesthetic action. In
Schwinn DA [ed]: Scientific Principles of Anesthesia, vol 2. Philadelphia, Current
Medicine, 1998.)
Figure 3-32
Cross-section of a schematized γ-aminobutyric acid
(GABA)-benzodiazepine receptor complex, which is a prototypical ligand-gated ion
channel complex. Binding of benzodiazepine agonists to the GABAA
ligand-gated
ion channel facilitates the action of endogenous GABA. This results in increased
inhibitory chloride ion flux in the central nervous system. A,
Schematic drawing of the GABAA
ligand-gated ion channel complex shows
the receptor sites for benzodiazepines and GABA, as well as distinct receptor sites
for barbiturates and alcohol. B, Model of the GABAA
receptor and chloride ion channel shows that the protein complex is composed of a
hetero-oligomer of five subunits, including α, β, γ, and δ
or ρ polypeptides. Each subunit has four putative membrane-spanning domains
(numbered 1 through 4 and represented by cylinders). (A,
from Berkowitz DE: Cellular signal transduction. In
Schwinn DA [ed]: Scientific Principles of Anesthesia, vol 2. Philadelphia, Current
Medicine, 1998; B, adapted from Firestone L, Quinlan
J, Gyulai F: Mechanisms of anesthetic action. In
Schwinn DA [ed]: Scientific Principles of Anesthesia, vol 2. Philadelphia, Current
Medicine, 1998.)
Another type of excitable membrane protein is the ion pump. The sodium-potassium-adenosine triphosphatase (ATPase) pump is perhaps the most familiar ion pump to the anesthesiologist because it is inhibited by digitalis. Extracellular fluid has high sodium and low potassium levels, whereas intracellular fluid has high potassium and low sodium concentrations. Because the nerve at rest is selectively permeable to potassium, but not to sodium, potassium moves extracellularly, creating a net positive extracellular charge and a net negative intracellular charge. Action potentials activate sodium channels, allowing sodium to rush intracellularly along chemical and electrical gradients. The sodium-potassium-ATPase pump then rapidly pumps sodium out of the cell in exchange for potassium, returning the cell to its original cation composition and electrical gradient. Drugs that act on ion pumps alter intracellular-extracellular cation ratios, resulting in altered membrane electrical potential.
Digitalis acts by inhibiting the sodium-potassium-ATPase pump. This is of special importance in the myocardial cell, where sodium-potassium-ATPase exchange is replaced by slower sodium-calcium exchange, thereby increasing intracellular calcium concentrations. Because calcium increases myocardial contractility, improved myocardial pump function results. This is the basis of digitalis use in the treatment of congestive heart failure.
The binding of a hormone or drug to its receptor does not instantly produce clinical effects. Instead, a series of rapid biochemical events couples receptor binding to ultimate clinical effects. These biochemical events are called second messengers. Because alterations in second messenger coupling can alter the effectiveness of a drug, it is important that the anesthesiologist understand the general principles of second messenger action.
Many membrane receptors couple to their primary second messenger through G proteins, which are intermediate regulatory molecules. The coupling of G proteins to receptor-hormone complexes requires energy in the form of guanosine triphosphate (GTP). The hydrolysis of G protein-associated GTP to GDP is regulated by another set of proteins, called RGS proteins. After the receptor interacts with the G protein, the biochemical reaction in the effector cascade is triggered. There are stimulating (e.g., Gs , Gq ) and inhibitory (e.g., Gi , Go ) G proteins, and the physiologic effect is determined by the specific G protein and subsequent cellular response. G proteins are heterotrimeric, composed of three subunits: α, β, and γ. Receptor and G protein interactions result in dissociation of the G protein subunits into α and βγ. The α-subunit of most G proteins confers specificity between receptor and effectors. Although βγ-subunits were originally thought simply to anchor G proteins to the cell membrane, dissociated βγ-subunits are clearly capable of directly stimulating second messengers. The βγ-subunits play a role in anchoring regulatory kinases to the cell membrane, leading to enhanced phosphorylation of membrane receptors.
One of the best understood second messenger systems is the adenylyl cyclase system. In this system, complexes of stimulatory G protein receptors and hormones increase activity of the enzyme adenylyl cyclase, resulting in increased levels of cyclic adenosine 3',5'-monophosphate (cAMP) in the cell ( Fig. 3-33 ). Inhibitory G protein-receptor-hormone complexes decrease the activity of adenylyl cyclase, resulting in decreased levels of cellular cAMP. In general, increased levels of intracellular cAMP activate protein kinases that phosphorylate various proteins, ion channels, and second messenger enzymes. For example, β-adrenergic-receptor stimulation results in stimulation of adenylyl cyclase activity, increasing production of intracellular cAMP, which activates protein kinase A, which phosphorylates the L-type calcium channel in myocardium to produce increases in intracellular calcium, ultimately leading to increased inotropy. Examples of drugs or hormones that activate adenylyl cyclase include glucagon and histamine. In contrast, muscarinic (M2 , M4 ) agonists, α2 -adrenergic agonists, and adenosine generally inhibit adenylyl cyclase.
Another example of a second messenger system is the phosphatidylinositol system. Hydrolysis of phosphatidyl-inositol-4,5-bisphosphate (PIP2 ) in the cell membrane, catalyzed by Gq activation of phospholipase C, generates two main second messenger molecules, inositol-1,4,5-trisphosphate (IP3 ) and 1,2-diacylglycerol (DAG). IP3 then mobilizes intracellular calcium from nonmitochondrial intracellular stores by interacting with distinct IP3 receptors on the surface of these organelles. The resulting increase in intracellular calcium levels produces biologic effects such as smooth muscle contraction. Examples of drugs that mediate phosphatidylinositol hydrolysis include α1 -adrenergic, endothelin, and muscarinic (M1 , M3 , M5 ) agonists.
Stimulation of receptors and second messengers ultimately leads to physiologic effects in a given tissue. The physiologic effects produced depend on the presence of
 Figure 3-33
Schematic drawing of the signal transduction cascade
of the β-adrenergic receptor (βAR). ATP, adenosine triphosphate; βγ,
stimulatory G protein βγ subunit; cAMP, cyclic adenosine monophosphate;
Gs
α, α-subunit of the stimulatory G protein (Gs
).
Figure 3-33
Schematic drawing of the signal transduction cascade
of the β-adrenergic receptor (βAR). ATP, adenosine triphosphate; βγ,
stimulatory G protein βγ subunit; cAMP, cyclic adenosine monophosphate;
Gs
α, α-subunit of the stimulatory G protein (Gs
).
 Figure 3-34
Adrenergic receptor subtype physiology. HR, heart rate;
NE, norepinephrine.
Figure 3-34
Adrenergic receptor subtype physiology. HR, heart rate;
NE, norepinephrine.
|
|
|
|
|
|
|
|
|
|
|
|
|