|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hypoxemia resulting from mechanical failure of the O2 supply system (also see Chapter 9 ) or the anesthesia machine is a recognized hazard of anesthesia. Disconnection of the patient from the O2 supply system (usually at the juncture of the endotracheal tube and the elbow connector) is by far the most common cause of mechanical failure to deliver O2 to the patient. Other reported causes of failure of the O2 supply during anesthesia include the following: an empty or depleted O2 cylinder, substitution of a nonoxygen cylinder at the O2 yoke because of absence or failure of the pin index, an erroneously filled O2 cylinder, insufficient opening of the O2 cylinder (which hinders free flow of gas as pressure decreases), failure of gas pressure in a piped O2 system, faulty locking of the piped O2 system to the anesthesia machine, inadvertent switching of the Schrader adapters on piped lines, crossing of piped lines during construction, failure of a reducing valve or gas manifold, inadvertent disturbance of the setting of the O2 flow meter, use of the fine O2 flow meter instead of the coarse flow meter, fractured or sticking
Esophageal intubation results in almost no ventilation. Virtually all other mechanical problems (except disconnection) with endotracheal tubes (such as kinking, blockage of secretions, and herniated or ruptured cuffs) cause an increase in airway resistance that may result in hypoventilation. Intubation of a main stem bronchus (also see Chapter 42 ) results in the absence of ventilation of the contralateral lung. Though potentially minimized by HPV, some perfusion to the contralateral lung always remains, and shunting increases and PaO2 decreases. A tube previously well positioned in the trachea may enter a bronchus after the patient or the patient's head is turned or moved into a new position.[136] Flexion of the head causes the tube to migrate deeper (caudad) into the trachea, whereas extension of the head causes cephalad (outward) migration of the endotracheal tube.[136] A high incidence of main stem bronchial intubation after the institution of a 30-degree Trendelenburg position has been reported. [137] Cephalad shift of the carina and mediastinum during the Trendelenburg position caused the previously "fixed" endotracheal tube to migrate into a main stem bronchus. Main stem bronchial intubation may obstruct the ipsilateral upper lobe in addition to the contralateral lung.[138] [139] Infrequently, the right upper bronchus or one of its segmental bronchi branches from the lateral wall of the trachea and may be occluded by a properly positioned endotracheal tube.
Patients under general anesthesia may have a reduced spontaneous VT for two reasons. First, increased work of breathing can occur during general anesthesia as a result of increased airway resistance and decreased CL. Airway resistance may be increased because of reduced FRC, endotracheal intubation, the presence of external breathing apparatus and circuitry, and possible airway obstruction in patients whose tracheas are not intubated.[140] [141] [142] CL is reduced as a result of some (or all) of the factors that can decrease FRC.[143] Second, patients may have a decreased drive to breathe spontaneously during general anesthesia (decreased chemical control of breathing) (see Fig. 17-31 ).
Decreased VT may cause hypoxemia
in two ways.[116]
First, shallow breathing may
promote atelectasis and cause a decrease in FRC (see the section "Ventilation Pattern").
[144]
[145]
Second,
decreased V̇E decreases the overall V̇A/![]() ratio of the lung, which decreases PaO2
(see Fig. 17-23
and Fig.
17-24
).[116]
This is likely to occur
with spontaneous ventilation during moderate to deep levels of anesthesia, in which
the chemical control of breathing is significantly altered.
ratio of the lung, which decreases PaO2
(see Fig. 17-23
and Fig.
17-24
).[116]
This is likely to occur
with spontaneous ventilation during moderate to deep levels of anesthesia, in which
the chemical control of breathing is significantly altered.
Hypocapnic alkalosis (hyperventilation) may result in decreased
PaO2
by several mechanisms: decreased
![]() T[102]
[103]
and increased V̇O2
[146]
[147]
(see the section "Decreased Cardiac Output
and Increased Oxygen Consumption"), a left-shifted oxy-Hb curve (see the section
"Oxygen-Hemoglobin Dissociation Curve"), decreased HPV[148]
(see the section "Inhibition of Hypoxic Pulmonary Vasoconstriction"), and increased
airway resistance and decreased compliance[149]
(see the section "Increased Airway Resistance").
T[102]
[103]
and increased V̇O2
[146]
[147]
(see the section "Decreased Cardiac Output
and Increased Oxygen Consumption"), a left-shifted oxy-Hb curve (see the section
"Oxygen-Hemoglobin Dissociation Curve"), decreased HPV[148]
(see the section "Inhibition of Hypoxic Pulmonary Vasoconstriction"), and increased
airway resistance and decreased compliance[149]
(see the section "Increased Airway Resistance").
Induction of general anesthesia is consistently accompanied by a significant (15% to 20%) decrease in FRC,[85] [94] [150] which usually causes a decrease in compliance.[143] The maximum decrease in FRC appears to occur within the first few minutes of anesthesia [85] [151] [152] and, in the absence of any other complicating factor, does not seem to decrease progressively during anesthesia. During anesthesia, the reduction in FRC is of the same order of magnitude whether ventilation is spontaneous or controlled. Conversely, in awake patients, FRC is only slightly reduced during controlled ventilation.[152] In obese patients, the reduction in FRC is far more pronounced than in normal patients, and the decrease is inversely related to the body mass index (BMI).[153] The reduction in FRC continues into the postoperative period.[154] For individual patients, the reduction in FRC correlates well with the increase in the alveolar-arterial PO2 gradient during anesthesia with spontaneous breathing,[155] during anesthesia with artificial ventilation,[152] and in the postoperative period.[154] The reduced FRC may be restored to normal or above normal by the application of PEEP.[84] [156] The following discussion considers all possible causes of reduced FRC.
Anesthesia and surgery are usually performed with the patient in the supine position (see Chapter 28 ). In changing from the upright to the supine position, FRC decreases by 0.5 to 1.0 L[85] [94] [150] because of a 4-cm cephalad displacement of the diaphragm by the abdominal viscera ( Fig. 17-33 ). Pulmonary vascular congestion may also contribute to the decrease in FRC in the supine position, particularly in patients who experienced orthopnea preoperatively. These FRC changes are magnified in obese patients, with the decrement directly related to BMI.[153]
At the end of a normal (awake) exhalation, there is slight tension in the inspiratory muscles and no tension in the expiratory muscles. Thus, at the end of a normal exhalation, there is a force tending to maintain lung volume and no force decreasing lung volume. After induction of general anesthesia, there is a loss of inspiratory tone and an appearance of end-expiratory tone in the abdominal expiratory muscles at the end of exhalation. The end-expiratory tone in the abdominal expiratory muscles increases intra-abdominal pressure, forces the diaphragm cephalad, and decreases FRC[151] [157] (see Fig. 17-33 ).
 Figure 17-33
Anesthesia and surgery may cause a progressive cephalad
displacement of the diaphragm. The sequence of events involves assumption of the
supine position, induction of anesthesia, establishment of paralysis, assumption
of several surgical positions, and displacement by retractors and packs. Cephalad
displacement of the diaphragm results in decreased functional residual capacity (↓
FRC). Pab, pressure of abdominal contents. (Redrawn with modification from
Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia, WB Saunders,
1995, Chapter 8.)
Figure 17-33
Anesthesia and surgery may cause a progressive cephalad
displacement of the diaphragm. The sequence of events involves assumption of the
supine position, induction of anesthesia, establishment of paralysis, assumption
of several surgical positions, and displacement by retractors and packs. Cephalad
displacement of the diaphragm results in decreased functional residual capacity (↓
FRC). Pab, pressure of abdominal contents. (Redrawn with modification from
Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia, WB Saunders,
1995, Chapter 8.)
With emphysema, exhalation may be accompanied by pursing the lips or grunting (partially closed larynx). An emphysematous patient exhales in either of these ways because both these maneuvers cause an expiratory retardation that produces PEEP in the intrathoracic air passage and decreases the possibility of airway closure and a decrease in FRC (see Fig. 17-17F ). Endotracheal intubation bypasses the lips and glottis and may abolish the normally present pursed-lip or grunting exhalation and in that way contributes to airway closure and loss of FRC in some spontaneously breathing patients.
In an upright subject, FRC and the position of the diaphragm are determined by the balance between lung elastic recoil pulling the diaphragm cephalad and the weight of the abdominal contents pulling it caudad.[159] There is no transdiaphragmatic pressure gradient.
The situation is more complex in the supine position. The diaphragm separates two compartments of markedly different hydrostatic gradients. On the thoracic side, pressure increases by approximately 0.25 cm H2 O/cm of lung height, [6] [7] and on the abdominal side, it increases by 1.0 cm H2 O/cm of abdominal height,[159] which means that in horizontal postures, progressively higher transdiaphragmatic pressure must be generated toward dependent parts of the diaphragm to keep the abdominal contents out of the thorax. In an unparalyzed patient, this tension is developed either by passive stretch and changes in shape of the diaphragm (causing an increased contractile force) or by neurally mediated active tension. With acute muscle paralysis, neither of these two mechanisms can operate, and a shift of the diaphragm to a more cephalad position occurs (see Fig. 17-33 ).[160] The latter position must express the true balance of forces on the diaphragm, unmodified by any passive or active muscle activity.
The cephalad shift in the FRC position of the diaphragm as a result of expiratory muscle tone during general anesthesia is equal to the shift observed during paralysis (awake or anesthetized patients).[151] [161] The equal shift suggests that the pressure on the diaphragm caused by an increase in expiratory muscle tone during general anesthesia is equal to the pressure on the diaphragm caused by the weight of the abdominal contents during paralysis. It is quite probable that the magnitude of these changes in FRC due to paralysis also depends on body habitus.
Induction of general anesthesia can result in increased expiratory muscle tone,[157] but the increased expiratory muscle tone is not coordinated and does not contribute to the exhaled volume of gas. In contrast, spontaneous ventilation during light general anesthesia usually results in a coordinated and moderately forceful active exhalation and larger exhaled volumes. Excessively inadequate anesthesia (relative to a given stimulus) results in very forceful active exhalation, which may produce exhaled volumes of gas equal to an awake expiratory vital capacity.
As during an awake expiratory vital capacity maneuver, forced expiration during anesthesia raises intrathoracic and alveolar pressure considerably above atmospheric pressure (see Fig. 17-17 ). This increase in pressure results in rapid outflow of gas, and because part of the expiratory resistance lies in the smaller air passages, a drop in pressure occurs between the alveoli and the main bronchi. Under these circumstances, intrathoracic pressure rises considerably above the pressure within the main bronchi. Collapse will occur if this reversed pressure gradient is sufficiently high to overcome the tethering effect of the surrounding parenchyma on the small intrathoracic bronchioles or the structural rigidity of cartilage in the large extrathoracic bronchi. Such collapse occurs in a normal subject during a maximal forced expiration and is responsible for the associated wheeze in both awake and anesthetized patients.[162]
In a paralyzed, anesthetized patient, the use of a subatmospheric expiratory pressure phase is analogous to a forced expiration in a conscious subject; the negative phase may set up the same adverse ΔP, which can cause airway closure, gas trapping, and a decrease in FRC. An excessively rapidly descending bellows of a ventilator during expiration has caused subatmospheric expiratory pressure and resulted in wheezing.[163]
The overall reduction in all components of lung volume during anesthesia results in reduced airway caliber, which increases airway resistance and any tendency toward airway collapse ( Fig. 17-34 ). The relationship between airway resistance and lung volume is well established ( Fig. 17-35 ).
 Figure 17-34
An anesthetized patient in the supine position has increased
airway resistance as a result of decreased functional residual capacity (FRC), decreased
caliber of the airways, endotracheal intubation, and connection of the endotracheal
tube (ET) to the external breathing apparatus and circuitry. (Redrawn with
modification from Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia,
WB Saunders, 1995, Chapter 8.)
Figure 17-34
An anesthetized patient in the supine position has increased
airway resistance as a result of decreased functional residual capacity (FRC), decreased
caliber of the airways, endotracheal intubation, and connection of the endotracheal
tube (ET) to the external breathing apparatus and circuitry. (Redrawn with
modification from Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia,
WB Saunders, 1995, Chapter 8.)
In addition to this expected increase in airway resistance in anesthetized patients, there are a number of additional special potential sites of increased airway resistance, including the endotracheal tube (if present), the upper and lower airway passages, and the external anesthesia apparatus. Endotracheal intubation reduces the
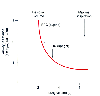 Figure 17-35
Airway resistance is an increasing hyperbolic function
of decreasing lung volume. Functional residual capacity (FRC) decreases when changing
from the upright to the supine position. (Redrawn with modification from
Lumb AB: Respiratory system resistance. In Lumb
AB [ed]: Nunn's Applied Respiratory Physiology, 5th ed. London, Butterworths, 2000,
p 67.)
Figure 17-35
Airway resistance is an increasing hyperbolic function
of decreasing lung volume. Functional residual capacity (FRC) decreases when changing
from the upright to the supine position. (Redrawn with modification from
Lumb AB: Respiratory system resistance. In Lumb
AB [ed]: Nunn's Applied Respiratory Physiology, 5th ed. London, Butterworths, 2000,
p 67.)
The respiratory apparatus often causes resistance that is considerably higher than the resistance in the normal human respiratory tract (see Fig. 17-34 ).[91] When certain resistors such as those shown in Figure 17-34 are joined in a series to form an anesthetic gas circuit, they generally add to produce larger resistance (as with resistance in series in an electrical circuit). The increase in resistance associated with commonly used breathing circuits and endotracheal tubes may impose an additional work of breathing that is two to three times normal.[142]
Patients undergoing anesthesia and surgery are often kept supine and immobile for long periods. Thus, some of the lung may be continually dependent and below the left atrium and therefore in zone 3 or 4 condition. Being in a dependent position, the lung is predisposed to accumulation of fluid. Coupled with excessive fluid administration, conditions sufficient to promote transudation of fluid into the lung are present and will result in pulmonary edema and decreased FRC. When mongrel dogs were placed in a lateral decubitus position and anesthetized for several hours ( Fig. 17-36, bottom horizontal axis ), expansion of the extracellular space with fluid (top horizontal axis) caused the PO2 (left-hand axis) of blood draining the dependent lung (closed circles) to
 Figure 17-36
Mongrel dogs anesthetized with pentobarbital (bottom
axis), placed in a lateral decubitus position, and subjected to progressive
extracellular fluid expansion (top axis) have a marked
decrease in the PO2
(left
vertical axis) of blood draining the dependent lung (solid
circles) and a smaller, much slower decrease in the PO2
of blood draining the nondependent lung (open circles).
The pulmonary arteriovenous shunt (right vertical axis)
rises progressively (triangles). (Redrawn
from Ray JF, Yost L, Moallem S, et al: Immobility, hypoxemia, and pulmonary arteriovenous
shunting. Arch Surg 109:537, 1974.)
Figure 17-36
Mongrel dogs anesthetized with pentobarbital (bottom
axis), placed in a lateral decubitus position, and subjected to progressive
extracellular fluid expansion (top axis) have a marked
decrease in the PO2
(left
vertical axis) of blood draining the dependent lung (solid
circles) and a smaller, much slower decrease in the PO2
of blood draining the nondependent lung (open circles).
The pulmonary arteriovenous shunt (right vertical axis)
rises progressively (triangles). (Redrawn
from Ray JF, Yost L, Moallem S, et al: Immobility, hypoxemia, and pulmonary arteriovenous
shunting. Arch Surg 109:537, 1974.)
General anesthesia is usually administered with an increased FIO2
.
In patients who have areas of moderately low V̇A/![]() ratios (0.1 to 0.01), administration of FIO2
greater than 0.3 adds enough O2
into the alveolar space in these areas
to eliminate the shuntlike effect that they have, and total measured right-to-left
shunting decreases. However, when patients with a significant amount of blood flow
perfusing lung units with very low V̇A/
ratios (0.1 to 0.01), administration of FIO2
greater than 0.3 adds enough O2
into the alveolar space in these areas
to eliminate the shuntlike effect that they have, and total measured right-to-left
shunting decreases. However, when patients with a significant amount of blood flow
perfusing lung units with very low V̇A/![]() ratios (0.01 to 0.0001) have a change in FIO2
from room air to 1.0, the very low V̇A/
ratios (0.01 to 0.0001) have a change in FIO2
from room air to 1.0, the very low V̇A/![]() units virtually disappear, and a moderately large right-to-left shunt appears.[16]
[17]
[166]
In these
studies, the increase in shunting was equal to the amount of blood flow previously
perfusing the areas with low V̇A/
units virtually disappear, and a moderately large right-to-left shunt appears.[16]
[17]
[166]
In these
studies, the increase in shunting was equal to the amount of blood flow previously
perfusing the areas with low V̇A/![]() ratios
during the breathing of air. Thus, in these studies the effect of breathing O2
was to convert units that had low V̇A/
ratios
during the breathing of air. Thus, in these studies the effect of breathing O2
was to convert units that had low V̇A/![]() ratios into shunt units. The pathologic basis for this data is the conversion of
low-V̇A/
ratios into shunt units. The pathologic basis for this data is the conversion of
low-V̇A/![]() units into atelectatic units.
units into atelectatic units.
The cause of the atelectatic shunting during O2
breathing
is presumably a large increase in O2
uptake by lung units with low V̇A/![]() ratios.[166]
[167]
A unit that has a low V̇A/
ratios.[166]
[167]
A unit that has a low V̇A/![]() ratio during
breathing of air will have a low PAO2
.
When an enriched O2
mixture is inspired, PAO2
rises, and the rate at which O2
moves from alveolar gas to capillary blood
increases greatly. The O2
flux may increase so much that the net flow
of gas into blood exceeds the inspired flow of gas, and the lung unit will become
progressively smaller. Collapse is most likely to occur if FIO2
is high, the V̇A/
ratio during
breathing of air will have a low PAO2
.
When an enriched O2
mixture is inspired, PAO2
rises, and the rate at which O2
moves from alveolar gas to capillary blood
increases greatly. The O2
flux may increase so much that the net flow
of gas into blood exceeds the inspired flow of gas, and the lung unit will become
progressively smaller. Collapse is most likely to occur if FIO2
is high, the V̇A/![]() ratio is low, the time
of exposure of the unit with low V̇A/
ratio is low, the time
of exposure of the unit with low V̇A/![]() to
high FIO2
is long, and Cv̄O2
is low. Thus, given the right V̇A/
to
high FIO2
is long, and Cv̄O2
is low. Thus, given the right V̇A/![]() ratio
and time of administration, an FIO2
as
low as 50% can produce absorption atelectasis.[166]
[167]
This phenomenon is of considerable significance
in the clinical situation for two reasons. First, enriched O2
mixtures
are often used therapeutically, and it is important to know whether this therapy
is causing atelectasis. Second, the amount of shunt is often estimated during breathing
of 100% O2
, and if this maneuver results in additional shunt, the measurement
will be hard to interpret.
ratio
and time of administration, an FIO2
as
low as 50% can produce absorption atelectasis.[166]
[167]
This phenomenon is of considerable significance
in the clinical situation for two reasons. First, enriched O2
mixtures
are often used therapeutically, and it is important to know whether this therapy
is causing atelectasis. Second, the amount of shunt is often estimated during breathing
of 100% O2
, and if this maneuver results in additional shunt, the measurement
will be hard to interpret.
In the supine position, the abdominal contents force the diaphragm cephalad and reduce FRC (see Chapter 28 ).[94] [151] [157] [161] The Trendelenburg position allows the abdominal contents to push the diaphragm further cephalad so that the diaphragm must not only ventilate the lungs but also lift the abdominal contents out of the thorax. The result is a predisposition to decreased FRC and atelectasis.[168] The Trendelenburg position-related decrease in FRC is exacerbated in obese patients.[153] Increased pulmonary blood volume and gravitational force on the mediastinal structures are additional factors that may decrease pulmonary compliance and FRC. In the steep Trendelenburg position, most of the lung may be below the left atrium and therefore in a zone 3 or 4 condition. As such, the lung may be susceptible to the development of pulmonary interstitial edema. Thus, patients with elevated Ppa, such as those with mitral stenosis, do not tolerate the Trendelenburg position well.[169]
In the lateral decubitus position, the dependent lung experiences a moderate decrease in FRC and is predisposed to atelectasis, whereas the nondependent lung may have increased FRC. The overall result is usually a slight to moderate increase in total-lung FRC.[170] The kidney and lithotomy positions also cause small decreases in FRC above that caused by the supine position. The prone position may increase FRC moderately.[170]
Rapid shallow breathing is often a regular feature of anesthesia. Monotonous shallow breathing may cause a decrease in FRC, promote atelectasis, and decrease compliance.[144] [145] [171] These changes with rapid shallow breathing are probably due to progressive increases in surface tension.[171] Initially, these changes may cause hypoxemia with normocapnia and may be prevented or reversed (or both) by periodic large mechanical inspirations, spontaneous sighs, PEEP, or a combination of these techniques.[171] [172] [173]
Tracheobronchial mucous glands and goblet cells produce mucus, which is swept by cilia up to the larynx, where it is swallowed or expectorated. This process clears inhaled organisms and particles from the lungs. The secreted mucus consists of a surface gel layer lying on top of a more liquid sol layer in which the cilia beat. The tips of the cilia propel the gel layer toward the larynx (upward) during the forward stroke. As the mucus streams upward and the total cross-sectional area of the airways diminishes, absorption takes place from the sol layer to maintain a constant depth of 5 mm.[174]
Poor systemic hydration and low inspired humidity reduce mucociliary flow by increasing the viscosity of secretions and slowing the ciliary beat.[175] [176] [177] Mucociliary flow varies directly with body or mucosal temperature (low inspired temperature) over a range of 32°C to 42°C.[178] [179] High FIO2 decreases mucociliary flow. [180] Inflation of an endotracheal tube cuff suppresses tracheal mucus velocity,[181] an effect that occurs within 1 hour, and apparently it does not matter whether
The mechanism for suppression of mucociliary clearance by the endotracheal tube cuff is speculative. In the report of Sackner and colleagues, [181] mucus velocity was decreased in the distal portion of the trachea, but the cuff was inflated in the proximal portion. Thus, the phenomenon cannot be attributed solely to damming of mucus at the cuff site. One possibility is that the endotracheal tube cuff caused a critical increase in the thickness of the layer of mucus proceeding distally from the cuff. Another possibility is that mechanical distention of the trachea by the endotracheal tube cuff initiated a neurogenic reflex arc that altered mucous secretions or the frequency of ciliary beating.
Other investigators have shown that when all the foregoing factors are controlled, halothane reversibly and progressively decreases, but does not stop mucus flow over an inspired concentration of 1 to 3 MAC.[182] The halothane-induced depression of mucociliary clearance was probably due to depression of the ciliary beat, an effect that caused slow clearance of mucus from the distal and peripheral airways. In support of this hypothesis is the finding that cilia are morphologically similar throughout the animal kingdom, and in clinical dosages, inhaled anesthetics, including halothane, have been found to cause reversible depression of the ciliary beat of protozoa.[183]
Decreased ![]() T in the presence
of constant O2
consumption (V̇O2
),
increased V̇O2
in the presence of
a constant
T in the presence
of constant O2
consumption (V̇O2
),
increased V̇O2
in the presence of
a constant ![]() T, or decreased
T, or decreased ![]() T
and increased V̇O2
must all result
in lower Cv̄O2
. Venous blood with
lowered Cv̄O2
then flows through
whichever shunt pathways exist, mixes with the oxygenated end-pulmonary capillary
blood, and lowers CaO2
(see Fig.
17-27
and Fig. 17-28
).
Figure 17-37
shows these
relationships quantitatively for
several different intrapulmonary shunts.[103]
[104]
The larger the intrapulmonary shunt, the greater the decrease in CaO2
because more venous blood with lower Cv̄O2
can admix with end-pulmonary capillary blood. Decreased
T
and increased V̇O2
must all result
in lower Cv̄O2
. Venous blood with
lowered Cv̄O2
then flows through
whichever shunt pathways exist, mixes with the oxygenated end-pulmonary capillary
blood, and lowers CaO2
(see Fig.
17-27
and Fig. 17-28
).
Figure 17-37
shows these
relationships quantitatively for
several different intrapulmonary shunts.[103]
[104]
The larger the intrapulmonary shunt, the greater the decrease in CaO2
because more venous blood with lower Cv̄O2
can admix with end-pulmonary capillary blood. Decreased ![]() T
may occur with myocardial failure and hypovolemia; the specific causes of these two
conditions are beyond the scope of this chapter. Increased V̇O2
may occur with excessive stimulation of the sympathetic nervous system, hyperthermia,
or shivering and can further contribute to impaired oxygenation of arterial blood.
[184]
T
may occur with myocardial failure and hypovolemia; the specific causes of these two
conditions are beyond the scope of this chapter. Increased V̇O2
may occur with excessive stimulation of the sympathetic nervous system, hyperthermia,
or shivering and can further contribute to impaired oxygenation of arterial blood.
[184]
Decreased regional PAO2 causes regional pulmonary vasoconstriction, which diverts blood flow away from hypoxic regions of the lung to better ventilated normoxic regions. The diversion of blood flow minimizes venous admixture from the underventilated or nonventilated lung regions. Inhibition of regional HPV could impair arterial oxygenation by permitting increased venous admixture from hypoxic or atelectatic areas of the lung (see Fig. 17-9 ).
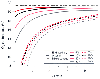 Figure 17-37
Effects of changes in cardiac output (
Figure 17-37
Effects of changes in cardiac output (![]() ) on the
O2
content of end-pulmonary capillary, arterial, and mixed venous blood
for a group of different transpulmonary right-to-left shunts. The magnitude of the
right-to-left shunt is indicated by the various numbered percent symbols for arterial
(solid line) and mixed venous (dashed
line) blood; the oxygen content of end-capillary blood is unaffected by
the degree of shunting. Note that a decrease in
) on the
O2
content of end-pulmonary capillary, arterial, and mixed venous blood
for a group of different transpulmonary right-to-left shunts. The magnitude of the
right-to-left shunt is indicated by the various numbered percent symbols for arterial
(solid line) and mixed venous (dashed
line) blood; the oxygen content of end-capillary blood is unaffected by
the degree of shunting. Note that a decrease in ![]() results in a greater decrease
in the arterial content of O2
the larger the shunt. (Redrawn
from Kelman GF, Nunn JF, Prys-Roberts C, et al: The influence of the cardiac output
on arterial oxygenation: A theoretical study. Br J Anaesth 39:450, 1967.)
results in a greater decrease
in the arterial content of O2
the larger the shunt. (Redrawn
from Kelman GF, Nunn JF, Prys-Roberts C, et al: The influence of the cardiac output
on arterial oxygenation: A theoretical study. Br J Anaesth 39:450, 1967.)
Because the pulmonary circulation is poorly endowed with smooth muscle, any condition that increases the pressure against which the vessels must constrict (i.e., Ppa) will decrease HPV. Numerous clinical conditions can increase Ppa and therefore decrease HPV. Mitral stenosis,[185] volume overload,[185] low (but greater than room air) FIO2 in nondiseased lung,[186] a progressive increase in the amount of diseased lung,[186] thromboembolism,[186] hypothermia,[187] and vasoactive drugs[188] can all increase Ppa. Direct vasodilating drugs (such as isoproterenol, nitroglycerin, and sodium nitroprusside), [61] [188] inhaled anesthetics,[189] and hypocapnia[148] [188] can directly decrease HPV. The selective application of PEEP to only the nondiseased lung can selectively increase PVR in the nondiseased lung and may divert blood flow back into the diseased lung.[190]
In the supine position, the weight of the abdominal contents pressing against the diaphragm is greatest in the dependent or posterior part of the diaphragm and least in the nondependent or anterior part of the diaphragm. In an awake patient breathing spontaneously, active tension in the diaphragm is capable of overcoming the weight of the abdominal contents, and the diaphragm moves the most in the posterior portion (because the posterior of the diaphragm is stretched higher into the chest, it has the smallest radius of curvature, and therefore it contracts most effectively) and least in the anterior portion. This circumstance is healthy because the greatest amount of ventilation occurs in areas with the most perfusion
Acute arterial hypoxemia from a transient right-to-left shunt through a patent foramen ovale (PFO) has been described, particularly during emergence from anesthesia.[193] However, unless a real-time technique of imaging the cardiac chambers is used (e.g., TEE with color flow Doppler imaging),[72] it is difficult to document an acute and transient right-to-left intracardiac shunt as a cause of arterial hypoxemia. Nonetheless, right-to-left shunting through a PFO has been described in virtually every conceivable clinical situation that afterloads the right side of the heart and increases right atrial pressure. When right-to-left shunting through a PFO is identified, administration of inhaled NO can decrease PVR and functionally close the PFO.[194]
In any given pulmonary disease, many of the mechanisms of hypoxemia listed earlier may be involved.[116] Pulmonary embolism (air, fat, thrombi) ( Fig. 17-38 ) (see Chapter 53 ) and the evolution of ARDS (see Chapter 75 ) ( Fig. 17-39 ) will be used to illustrate this point. A significant pulmonary embolus can cause severe increases in pulmonary artery pressure, and these increases can result in right-to-left transpulmonary shunting through opened arteriovenous anastomoses and the foramen ovale (possible in 20% of patients), pulmonary edema in nonembolized regions of the lung, and inhibition of HPV. The embolus may cause hypoventilation via increased dead space ventilation. If the embolus contains platelets, serotonin may be released, and such release can cause hypoventilation as a result of bronchoconstriction and pulmonary edema as a result of increased pulmonary capillary permeability. Finally, the pulmonary embolus can increase PVR (by platelet-induced serotonin release,[26] among other etiologies) and decrease cardiac output.
After major hypotension, shock, blood loss, sepsis, and other conditions, noncardiogenic pulmonary edema may occur and lead to acute respiratory failure or ARDS[195] (described more fully in Chapter 74 and Chapter 75 ). The syndrome can evolve during and after anesthesia and has the hallmark characteristics of decreased FRC and compliance and hypoxemia. After shock and trauma, plasma levels of serotonin, histamine, kinins, lysozymes, reactive oxygen species, fibrin degradation products, products of complement metabolism, and fatty acids increase. Sepsis
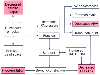 Figure 17-38
Mechanisms of hypoxemia during pulmonary embolism. See
the text for an explanation of the pathophysiologic flow diagram. AV, arteriovenous;
CAP PERM, capillary permeability; CC, closing capacity; FRC, functional residual
capacity; HPV, hypoxic pulmonary vasoconstriction; PA, pulmonary artery. (Redrawn
with modification from Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia,
WB Saunders, 1995, Chapter 8.)
Figure 17-38
Mechanisms of hypoxemia during pulmonary embolism. See
the text for an explanation of the pathophysiologic flow diagram. AV, arteriovenous;
CAP PERM, capillary permeability; CC, closing capacity; FRC, functional residual
capacity; HPV, hypoxic pulmonary vasoconstriction; PA, pulmonary artery. (Redrawn
with modification from Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia,
WB Saunders, 1995, Chapter 8.)
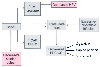 Figure 17-39
Mechanisms of hypoxemia during adult respiratory distress
syndrome. See the text for an explanation of the pathophysiologic flow diagram.
CAP PERM, capillary permeability; CC, closing capacity; FRC, functional residual
capacity; HPV, hypoxic pulmonary vasoconstriction; PA, pulmonary artery. (Redrawn
with modification from Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia,
WB Saunders, 1995, Chapter 8.)
Figure 17-39
Mechanisms of hypoxemia during adult respiratory distress
syndrome. See the text for an explanation of the pathophysiologic flow diagram.
CAP PERM, capillary permeability; CC, closing capacity; FRC, functional residual
capacity; HPV, hypoxic pulmonary vasoconstriction; PA, pulmonary artery. (Redrawn
with modification from Benumof JL: Anesthesia for Thoracic Surgery, 2nd ed. Philadelphia,
WB Saunders, 1995, Chapter 8.)
|
|
|
|
|
|
|
|
|
|
|
|
|