|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The lipophilic characteristics of drugs that promote their passage through biologic membranes hinder their excretion from the body. Drug metabolism is therefore essential for elimination and termination of biologic activity of lipophilic drugs because it usually results in the formation of more hydrophilic products that are less pharmacologically active and more readily excreted from the body ( Fig. 8-1 ). Drug metabolism sometimes results in the formation of intermediates whose pharmacologic or toxicologic activity are equal to, greater than, or different from the parent drug.[1] Unlike most drugs, the inhaled anesthetics are administered in great excess of the amount metabolized and are excreted mainly by exhalation. Biotransformation has little effect on the pharmacologic activity of anesthetics, but it may have a significant effect
 Figure 8-1
Effects of drug metabolism on excretion. Lipophilic
(fat-soluble) drugs are metabolized to form relatively more hydrophilic (water-soluble)
metabolites than the parent drug, and these metabolites are more easily excreted.
(From Weinshilboum R: Inheritance and drug response. N Engl J Med 348:529–537,
2003.)
Figure 8-1
Effects of drug metabolism on excretion. Lipophilic
(fat-soluble) drugs are metabolized to form relatively more hydrophilic (water-soluble)
metabolites than the parent drug, and these metabolites are more easily excreted.
(From Weinshilboum R: Inheritance and drug response. N Engl J Med 348:529–537,
2003.)
The liver is the primary organ of drug metabolism because of its large size, rich concentration of drug-metabolizing enzymes, and unique double blood supply, consisting of 70% of the flow from the portal vein and 30% from the hepatic artery. Blood in the portal vein comes from the alimentary tract, pancreas, and spleen. Toxic materials absorbed from the alimentary tract are processed by the liver before they enter the systemic circulation. Blood flows through the hepatic sinusoids from the periphery of the hepatic lobule, fed by portal veins and hepatic arteries, to the centrally located hepatic venule or central vein. The hepatocytes, which are bathed by the sinusoidal blood supply, are the major drug-metabolizing cells in the liver and body. Several drug-metabolizing enzyme systems are located within the smooth endoplasmic reticulum of hepatocytes, and others are found in the cytosol of these cells. In addition to the liver, organs with significant drug-metabolizing activity are the gastrointestinal tract, kidneys, and lungs.[4] [5]
The major enzymatic reactions biotransforming drugs into metabolites are oxidation, hydrolysis, and conjugation. It is common for a drug to be metabolized into several metabolites. The ratios of various metabolites of a drug depend on enzymatic reaction rates, drug concentration
The phase 1 enzymes most relevant to the metabolism of inhaled anesthetics are the cytochromes P450 (CYPs). These enzymes are capable of catalyzing several different types of oxidations reactions, including dehalogenations, N- and O-dealkylations, N- and S-oxidations, and deamination reactions. All of these reactions require CYP, oxygen, and NADPH-dependent cytochrome P450 reductase for activity and occur in the endoplasmic reticulum of cells, particularly hepatocytes. Under conditions of low oxygen tension, these enzymes may catalyze reductive reactions. Approximately 50 of the more than 1000 CYPs are functionally active in humans and are categorized into 17 families and subfamilies. Sequences with greater than 40% homology belong to the same family and are identified by an arabic numeral. The sequences of subfamily members are at least 55% identical and are distinguished by a capital letter. An arabic number is used to distinguish individual isoforms within a subfamily. CYP2E1, for example, is a member of the CYP2 family, which is a large family of isozymes that metabolizes many diverse drugs and endogenous compounds. [6] Approximately 10 isoforms in the CYP1, CYP2, and CYP3 families are responsible for most drug metabolism in humans ( Fig. 8-2 ). Most CYP isoforms metabolize multiple drugs, and some overlap in substrate specificity can be seen between the various isoforms. Two or more CYP isoforms may be involved in the overall metabolism of a drug. In human liver, CYP3A4 and CYP3A5 subfamilies account for as much as 60% of the total CYP present. CYP2E1 is particularly important in the oxidative metabolism of halogenated inhaled anesthetics, and like many CYPs, it is concentrated in the perivenular hepatocytes.
In phase 2 reactions, a polar molecule such as glucuronic acid, sulfate, or glycine is conjugated to a drug or its metabolites to produce highly hydrophilic products that are readily excreted into the urine or, in some cases, into the bile and gastrointestinal tract. Glucuronidation is perhaps the most important conjugation reaction, and uridine 5'-diphosphate glucuronosyltransferase (UGT) catalyzes the transfer of glucuronic acid to aromatic and aliphatic alchols, carboxylic acids, amines, and free sulfhydryl groups to form O-, N-, and S-glucuronides, respectively. UGTs, like CYPs, exist as multiple isoforms with different substrate specificities and are localized in the
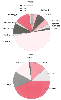 Figure 8-2
Proportion of drugs metabolized by major phase 1 or phase
2 enzymes. The relative size of each pie section indicates the estimated percentage
of phase 1 (left) or phase 2 (right)
metabolism that each enzyme contributes to the metabolism of drugs based on literature
reports. Enzymes that have functional allelic variants are indicated by an asterisk.
In many cases, more than one enzyme is involved in a particular drug's metabolism.
CYP, cytochrome P450; DPYD, dihydropyridine dehydrogenase; GST, glutathione S-transferase;
NAT, N-acetyltransferase; ST, sulfotransferase; TPMT,
thiopurine methyltransferase; UGT, UDP-glucuronosyltransferase. (Adapted
from Wilkinson G: Pharmacokinetics: The dynamics of drug absorption, distribution
and elimination. In Hardman JG, Limbird LE, Goodman
GA [eds]: Goodman and Gilman's the Pharmacological Basis of Therapeutics, 10th ed.
New York, McGraw-Hill, 2001.)
Figure 8-2
Proportion of drugs metabolized by major phase 1 or phase
2 enzymes. The relative size of each pie section indicates the estimated percentage
of phase 1 (left) or phase 2 (right)
metabolism that each enzyme contributes to the metabolism of drugs based on literature
reports. Enzymes that have functional allelic variants are indicated by an asterisk.
In many cases, more than one enzyme is involved in a particular drug's metabolism.
CYP, cytochrome P450; DPYD, dihydropyridine dehydrogenase; GST, glutathione S-transferase;
NAT, N-acetyltransferase; ST, sulfotransferase; TPMT,
thiopurine methyltransferase; UGT, UDP-glucuronosyltransferase. (Adapted
from Wilkinson G: Pharmacokinetics: The dynamics of drug absorption, distribution
and elimination. In Hardman JG, Limbird LE, Goodman
GA [eds]: Goodman and Gilman's the Pharmacological Basis of Therapeutics, 10th ed.
New York, McGraw-Hill, 2001.)
Various factors may affect drug metabolism, including environmental determinants, disease, age, gender, and genetics. The activity of many drug-metabolizing enzymes may be increased (i.e., induced) or decreased (i.e., inhibited) by concomitantly administered drugs. Enzyme induction results from enhanced gene transcription after prolonged exposure to an induction agent or, in some instances, to a decreased rate of enzyme degradation. The induction of CYPs by numerous drugs and environmental agents has been extensively studied by many researchers.[7] Enzyme inducers are often highly lipophilic drugs and environmental chemicals that are metabolized by the CYP isozymes they induce. The inducing properties of a drug are unrelated to the nature of its pharmacologic or toxicologic activity and may vary markedly from those of other drugs in the same class. Induction by drugs such as phenobarbital results in proliferation of the smooth endoplasmic reticulum and an increase in liver weight. With this type of inducer, NADPH-cytochrome P450 reductase and specific CYP isozymes are preferentially increased. Although other inducers increase the synthesis of specific CYP isozymes, they do not affect cytochrome P450 reductase or liver weight. Many classes of drugs and environmental chemicals, including anesthetics, anticonvulsants, insecticides, sedatives, steroids, and tranquilizers, contain one or more compounds considered to be enzyme inducers.[7] [8] Even the inhaled anesthetics can induce drug-metabolizing enzymes if exposure is sufficiently prolonged.[9] [10] If the parent compound is toxic, enhanced metabolism may decrease toxicity. In contrast, when metabolites are more toxic than the parent compound, metabolism increases toxicity. In many cases involving induction, the dosage of an affected drug must be increased to maintain the therapeutic effect.
Enzyme-inducing agents have the potential to modify acute and chronic toxicities of anesthetics. In view of the current practice of polypharmacy, enzyme induction may be more common in patients undergoing surgery than previously appreciated. Enzyme induction does not necessarily increase the metabolism of all drugs from the same class. For example, unlike methoxyflurane metabolism, enflurane metabolism is not significantly increased in vivo after phenobarbital or phenytoin treatment in humans.[11] Even when anesthetics that are metabolized to toxic metabolites are administered to surgical patients taking enzyme-inducing drugs, significant amounts of toxic metabolites are not necessarily produced.[12] [13]
The consequences of enzyme inhibition for therapeutic activity and toxicity can be just as great as those of enzyme induction. This can lead to an increase in the plasma concentration of the parent drug and a reduction in drug metabolites, as well as exaggerated and prolonged pharmacologic effects and an increased chance of toxicity. Many compounds inhibit the activity of the drug-metabolizing enzymes and thereby alter the duration and intensity of pharmacologic action and the severity of toxic effects. There are several mechanisms of inhibition.[14] Protein synthesis inhibitors such as cycloheximide decrease enzyme synthesis and reduce enzyme concentrations. Other agents are reversible inhibitors that compete for the active site of the same enzyme responsible for metabolism of the drug of interest. Still others are irreversible inhibitors that degrade the heme in cytochrome P450. Inhibition of CYP3A is common and important because of the increased expression of CYP3A in the intestinal epithelium and the fact that oral ingestion is the most common route of entry for drugs. There is a potential increase in bioavailability associated with a reduction of first-pass metabolism. Potent CYP3A inhibitors include ritonavir, diltiazem, nicardipine, and verapamil. More general inhibitors of CYPs include aminodarone and cimetidine.
Some sex-related differences in drug-metabolizing activities have been observed, particularly for CYP3A, but such differences are minor relative to previously described factors. An exception is the treatment of seizure patients with phenytoin during pregnancy. Because the liver is the major site of drug metabolism, intrinsic liver disease leading to hepatic dysfunction can impair drug metabolism. Diseases such as hepatitis, primary biliary cirrhosis, alcoholic liver disease, cirrhosis, and hepatocarcinoma can lead to a 50% impairment of enzymatic activity. Severe cardiac failure with decreased hepatic perfusion and impaired metabolism can also occur, as evidenced by the twofold difference in the loading and maintenance doses of lidocaine for dysrhythmia treatment in patients with heart failure. Moreover, viral infections can lead to the inhibition of CYP-mediated reactions.
Phase 1 and phase 2 enzymes begin to mature during the postnatal period, but the levels may be low enough in some case to result in toxicities if drug therapy is not carefully monitored (see Chapter 60 ). For example, the impairment of bilirubin glucuronidation at birth can lead to hyperbilirubinemia of the newborn and "gray baby" syndrome caused by high levels of the antibiotic chloramphenicol.[15] [16] As children age, phase 1 enzymes appear to be affected to a greater degree than phase 2 enzymes. The predominant isoform expressed in fetal liver is CYP3A7, which peaks shortly after birth and then declines rapidly to levels that are undetectable in most adults.[17] Within hours of birth, CYP2E1 activity increases, followed by CYP2D6, CYP3A4, CYP2C9, and CYP2C19. CYP1A2 is the last isoform to appear at about 2 months of age. CYP2C9 and CYP2C19 are responsible for the biotransformation of phenytoin. The apparent half-life of phenytoin is prolonged to about 75 hours in preterm neonates but decreases to about 20 hours in term neonates during the first week of life and 8 hours by the second week of life.[18]
|
|
|
|
|
|
|
|
|
|
|
|
|