|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
This section deals with the effect of anesthetics on CBF and CMR. It includes limited mention of influences on autoregulation, CO2 responsiveness, and CBV. Discussions of effects on CSF dynamics, the BBB, and epileptogenesis are presented in the later section "Cerebral Physiology in Pathologic States."
In neuroanesthesia, considerable emphasis is placed on the manner in which anesthetic drugs and techniques influence CBF. The rationale is twofold. First, delivery of energy substrates is dependent on CBF, and in the setting of ischemia, modest alterations in CBF can substantially influence neuronal outcome. Second, control and manipulation of CBF are central to the management of ICP because as CBF varies in response to vasoconstrictor-vasodilator influences (e.g., PaCO2 and volatile anesthetics), CBV varies linearly with it, albeit with some variation among anesthetics in the CBF-CBV ratio.[136] In normal brain, CBV is approximately 5 mL/100 g of brain tissue,[137] and over a PaCO2 range of approximately 25 to 70 mm Hg, CBV changes by about 0.049 mL/100 g for each 1-mm Hg change in PaCO2 . [138] [139] In an adult brain weighing about 1400 g, this can amount to a 20-mL change in total CBV for a PaCO2 range of 25 to 55 mm Hg.
Although CBV and CBF usually vary in parallel, exceptions do occur. CBV increases during cerebral ischemia,[140] [141] and it may vary independently of CBF when MAP is the manipulated variable. Autoregulation normally serves to prevent MAP-related increases in CBV. In fact, as cerebral circulation constricts to maintain CBF constant in the face of rising MAP, CBV actually decreases. [142] When autoregulation is impaired or its upper limit (≅150 mm Hg) is exceeded, CBF and CBV then increase in parallel as arterial pressure rises (see Fig. 21-4 ). Declining MAP results in a progressive increase in CBV as the cerebral circulation dilates to maintain constant flow, and exaggerated increases in CBV occur as MAP falls below the LLA.[142] In normal subjects, the initial increases in CBV do not result in significant elevation of ICP because there is latitude for compensatory adjustments by other intracranial compartments (e.g., translocation of venous blood and CSF to extracerebral vessels and the spinal CSF space, respectively). When intracranial compliance * is reduced, an increase in CBV can cause herniation or reduce CPP sufficiently to cause ischemia.
Several investigations on the effects of anesthetics on CBV in normal brain have been conducted.[144] [145] [146] In general, the effects observed confirm a parallel relationship between CBF and CBV. However, the relationship is not consistently 1:1,[136] [147] and CBF-independent influences on CBV may occur. It is also an unexplored possibility that anesthetics may influence the venous side of the cerebral circulation. Although the intracranial veins are a largely passive compartment, some evidence indicates that in certain species, there is some active control of venous caliber by either neurogenic or humoral mechanisms.[75] [77] [148]
The general pattern of the effect of intravenous anesthetics is one of parallel alterations in CMR and CBF. The vast majority of intravenous anesthetics cause a reduction in both. Ketamine, which causes an increase in CMR and CBF, is the exception. The effects of selected intravenous anesthetics on human CBF are compared in Figure 21-6 .
It is probable that intravenous anesthetic-induced changes in CBF are largely the result of effects on CMR with parallel (coupled) changes in CBF. If this were the entire explanation, the CBF/CMR ratio would be the same for all anesthetics, but it is not. It is therefore likely that there are also direct effects on cerebral vascular smooth muscle (e.g., vasoconstriction, vasodilation, alteration of autoregulatory function) that make contributions to the net effect. For instance, although barbiturates are generally thought of as cerebral vasoconstrictors, some barbiturates actually cause relaxation of cerebral vascular smooth muscle in isolated vessel preparations.[161] [162] [163] However, a substantial reduction in CMR occurs in vivo, and the net effect at the point of EEG suppression is vasoconstriction and a substantial decrease in CBF.[21] It appears that in general, autoregulation and CO2 responsiveness are preserved during administration of intravenous anesthetics.
A dose-dependent reduction in CBF and CMR occurs with barbiturates. With the onset of anesthesia, CBF and CMRO2 are reduced by about 30%. [164] [165] When large doses of thiopental cause complete EEG suppression, CBF and CMR are reduced by about 50%.[21] [149] [166] Further increases in the dose of barbiturate have no additional effect on CMR.[21] [167] These observations suggest that the major effect of nontoxic doses of depressant anesthetics is a reduction in the component of cerebral metabolism that is linked to electrical brain function (e.g., neurophysiologic activity), with only minimal effects on the second component, that related to cellular homeostasis (see Fig. 21-1 ).
Tolerance to the CBF/CMR effects of barbiturates may develop quickly. [168] [169] In patients with severe head injury in whom "barbiturate coma" was maintained for 72 hours, the thiamylal blood concentration required to maintain EEG burst suppression was observed to be increased by the end of the first 24 hours and continued to increase over the next 48 hours.[170] During deep pentobarbital anesthesia, autoregulation is maintained to arterial pressures as low as 60 mm Hg.[171] CO2 responsiveness also persists.[172]
The effects of propofol (2,6-diisopropylphenol) on CBF and CMR appear to be quite similar to those of the barbiturates. Three
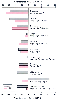 Figure 21-6
Changes in cerebral blood flow (CBF) and the cerebral
metabolic rate of oxygen (CMRO2
) caused by intravenous anesthetic agents.
The data are derived from human investigations and are presented as the percent
change from unanesthetized control values. Dexmedetomidine CMR values were determined
on a background of 0.5% isoflurane anesthesia. See the text for details. No data
for the CMRO2
effects of midazolam in humans are available. (Data
from references [149]
[150]
[151]
[152]
[153]
[154]
[155]
[156]
[157]
[158]
[159]
[160]
.)
Figure 21-6
Changes in cerebral blood flow (CBF) and the cerebral
metabolic rate of oxygen (CMRO2
) caused by intravenous anesthetic agents.
The data are derived from human investigations and are presented as the percent
change from unanesthetized control values. Dexmedetomidine CMR values were determined
on a background of 0.5% isoflurane anesthesia. See the text for details. No data
for the CMRO2
effects of midazolam in humans are available. (Data
from references [149]
[150]
[151]
[152]
[153]
[154]
[155]
[156]
[157]
[158]
[159]
[160]
.)
Both CO2 responsiveness and autoregulation appear to be preserved during the administration of propofol in humans,[178] [179] even when propofol is administered in doses that produce burst suppression of the EEG.[180] The magnitude of the reduction in CBF during hypocapnia is decreased during propofol administration. This effect is probably due to CMR suppression-induced cerebral vasoconstriction, thereby limiting further hypocapniamediated vasoconstriction. Seizures and opisthotonos have been reported to occur after propofol anesthesia. [181] [182] [183] [184] [185] However, systematic studies in both humans[186] [187] [188] and animals,[189] [190] though identifying the occurrence of occasional dystonic and choreiform[186] movements, have failed to confirm the notion that propofol is proconvulsant. In fact, propofol appears to be anticonvulsant in animals (mice).[189] [190] Furthermore, ECT seizures were shorter after induction with propofol than with methohexital, [191] which is more consistent with an anticonvulsant effect. In addition, propofol sedation has been widely used during "awake" resection of seizure foci and other intracranial lesions.[187] [188] [192] Although pronounced high-amplitude beta-frequency activity in the EEG has been observed,[193] there has not been an unexpected incidence of seizures.
The effects of etomidate on CBF and CMR are also superficially similar to those of barbiturates. Roughly parallel reductions in CBF and CMR occur in humans[151] [194] and are in general accompanied by progressive suppression of the EEG.[195] Induction of anesthesia with either thiopental or etomidate resulted in a similar reduction in MCA flow velocity of about 27%.[196] The CBF/CMR changes are substantial. Renou and coworkers[151] gave approximately 0.2 mg/kg of etomidate to adults and observed mean reductions in CBF and CMR of 34% and 45%, respectively. In dogs, as is the case with barbiturates, no further reduction in CMR occurs when additional drug is administered beyond a dose sufficient to produce EEG suppression.[26] This latter phenomenon has not been demonstrated in humans. However, Bingham and colleagues[197] observed that etomidate lowered ICP when administered to severely head injured patients in whom EEG activity was well preserved, but it was ineffective when there was substantial antecedent EEG suppression. Canine data reviewed by Milde and colleagues[26] suggest that though substantial, the global CMR suppression attainable with etomidate is slightly less profound than that achieved with isoflurane and barbiturates. These data are consistent with the observations of Davis and associates[198] that unlike barbiturates, which cause CMR suppression throughout the brain, the CMR suppression caused by etomidate is regionally variable and occurs predominantly in the forebrain structures.
Etomidate has been shown to be effective in reducing ICP without causing a reduction in CPP in patients with intracranial tumors[199] and head-injured patients.[200] However, etomidate administration has been shown to result in an exacerbation of brain tissue hypoxia and acidosis in patients in whom the MCA was temporarily occluded during surgery. [201] Additional concerns regarding the occurrence of adrenocortical suppression and renal injury caused by the propylene glycol vehicle [202] will probably preclude more than episodic use.
Reactivity to CO2 is preserved in humans during etomidate administration.[151] [195] Autoregulation has not been evaluated. Myoclonus and epileptogenesis are discussed in a later section.
Despite inconsistencies in the available information, it is likely that narcotics have relatively little effect on CBF and CMR in the normal, unstimulated nervous system.[203] When changes occur, the general pattern is one of modest reductions in both CBF and CMR. The inconsistencies in the literature probably arise largely because in many studies, the "control" states entailed paralysis and nominal sedation, often with nitrous oxide alone. In these studies, in which substantial reductions in CBF and CMR were frequently observed, the effect of the narcotic was probably a combination of the inherent effect of the drug plus a substantial component attributable to reduction in arousal. Comparable effects related to reduction of arousal may occur and be important clinically. However, they should be viewed as nonspecific effects of sedation or pain control, or both, rather than specific properties of narcotics. The following discussion emphasizes investigations in which control measurements were unlikely to have been markedly influenced by arousal phenomena.
When morphine (≅1 mg/kg) was administered as the sole agent in humans, Moyer and colleagues[204] observed no effect on global CBF and a 41% decrease in CMRO2 . The latter is a substantial reduction, and the absence of a simultaneous CBF adjustment is surprising. No other investigations of morphine alone in humans have been conducted. Jobes and coworkers gave morphine (1 and 3 mg/kg) with 70% N2 O to patients and observed no significant change in CBF or CMR.[152] The N2 O that was used might be expected to have caused a tendency toward increases in CBF and CMR. The relative absence of net changes in these variables from the awake control measurements suggests a small to moderate depressive effect of morphine on CBF and CMR at this large dose. Takeshita and associates[205] gave morphine, 3 mg/kg, to dogs sedated with 70% N2 O (≅0.6 MAC). They observed reductions in global CBF and CMRO2 of approximately 60% and 17%, respectively. These data cannot be completely reconciled, and the data of Jobes and colleagues[152] (presented earlier) are probably relevant to the
Autoregulation was observed to be intact between MAP values of 60 and 120 mm Hg in human volunteers anesthetized with morphine, 2 mg/kg, and 70% N2 O.[206]
Limited human data are available. Vernhiet and colleagues[153] measured CBF and CMRO2 before and during anesthesia with 12 to 30 (mean, 16) µg/kg of fentanyl plus 50% N2 O in patients about to undergo cerebral angiography. Atropine and pancuronium were the only other drugs administered. Neither CBF nor CMRO2 changed significantly from awake control values in their group of six subjects. However, one of the patients (an epileptic with a normal computed tomography [CT] scan) had dramatic and unexplained increases in both CBF and CMRO2 . For the remaining five, CBF and CMRO2 decreased by 21% and 26%, respectively (P < .05). The data for fentanyl/N2 O presented in Figure 21-6 are derived from these five patients who received an average of 17 µg/kg of fentanyl. Murkin and coworkers measured CBF before and after induction of anesthesia with high-dose fentanyl, 100 µg/kg, and diazepam, 0.4 mg/kg. [207] CBF fell by 25%, although part of this effect may well have been the result of the benzodiazepine (see later) rather than fentanyl. Firestone and associates, using PET, observed a heterogeneous CBF response to 1.5 µg/kg of fentanyl in healthy volunteers. Increases occurred in the frontal, temporal, and cerebellar areas simultaneous with decreases in discreet areas associated with painrelated processing.[208] Additional data have been derived from animal experiments. McPherson and Traystman[209] administered fentanyl (25 µg/kg) to pentobarbital-anesthetized dogs and observed no effect on CBF and CMR. CO2 responsiveness and autoregulation were unaffected, and the hyperemic CBF response to hypoxia also remained intact. Several investigations in lightly anesthetized animals[210] [211] [212] have demonstrated much larger fentanyl-induced reductions in CBF or CMR (or both) than those observed in humans.
These data taken together suggest that fentanyl will cause a moderate global reduction in CBF and CMR in the normal quiescent brain and will, like morphine, cause larger reductions when administered during arousal.
McPherson and colleagues[213] administered alfentanil, 320 µg/kg, to pentobarbitalanesthetized dogs. They observed no changes in CBF, CMR, CO2 responsiveness, autoregulation, or the CBF response to hypoxia. No studies of the CMR effects of alfentanil in humans have been performed. Schregel and coworkers administered 25 to 50 µg/kg of alfentanil to patients receiving 60% N2 O after induction of anesthesia with thiopental.[214] CBFV decreased transiently. A Doppler measure of MCA diameter was simultaneously unchanged, thus suggesting that the CBFV reduction was indicative of a decrease in CBF. Mayberg and colleagues also observed no changes in CBFV in response to 25 to 50 µg/kg of alfentanil given to patients during maintenance of anesthesia with isoflurane-N2 O. [215]
Although some laboratory investigations have revealed apparent sufentanil-induced increases in CBF and CMR,[216] most investigations in both animals[211] [217] [218] and humans indicate that sufentanil causes, depending on the dose, either no change or reductions in CBF and CMR. Stephan and associates[154] measured CBF and CMRO2 in patients before and after induction of anesthesia with 10 µg/kg of sufentanil. They observed a 29% reduction in CBF and a 22% reduction in CMRO2 . Murkin and colleagues, in a study involving the same dose of sufentanil and a similar design, made essentially identical observations.[219] Mayer and coworkers gave 0.5 µg/kg of sufentanil to volunteers and observed no change in CBF.[220] Weinstabl and colleagues observed reductions in CBFV when 1.0 and 2.0 µg/kg of sufentanil were given to intensive care unit (ICU) patients with increased ICP.[221] Neither Weinstabl and colleagues[221] nor Mayer and coworkers,[220] who administered sufentanil to healthy volunteers, observed changes in CBF velocity after 0.5 µg/kg of sufentanil.
The data from the aforementioned studies lead to the anticipation of no change or a reduction in ICP as a result of the administration of either sufentanil or alfentanil. With respect to sufentanil, the bulk of the data derived from animals [216] [217] [218] [222] and humans[221] [223] [224] [225] [226] have revealed no change in ICP after its administration. However, in some investigations in humans, sufentanil was associated with modest increases in neuraxis pressure.[227] [228] [229] Subsequent investigations appear to indicate that the ICP increases associated with sufentanil are at least in large part the consequence of a normal autoregulatory response to the sudden MAP reduction that can occur as a result of sufentanil administration.[230] The "message" for the clinician should probably be that sufentanil (and for that matter, fentanyl as well[225] ) are best administered in a manner that does not produce a sudden reduction in MAP. MAP reduction will clearly reduce CPP and may increase ICP, each of which, when sufficiently extreme, may be deleterious. However, it should be noted that the ICP increases attributed to sufentanil have been small. Furthermore, four investigations[231] [232] [233] [234] that compared conditions in the surgical field, including pressure under brain retractors, [231] identified no adverse influences attributable to sufentanil. Accordingly, sufentanil need not be viewed as contraindicated in any way, although it should be used with attention to its effect on MAP.
Although fewer data are available, the general pattern is similar and the conclusions should be the same as for sufentanil (preceding paragraph).[227] [235] [236] [237] [238] Alfentanil was included with fentanyl and sufentanil in two of the investigations of conditions in the surgical field mentioned in connection with sufentanil.[231] [232] No adverse effects were noted.
Investigations of moderate doses of remifentanil in patients have revealed minimal effects that are very similar to those of the other synthetic narcotics (with the exception of its substantially shorter duration of action). In patients undergoing craniotomy for supratentorial space-occupying lesions, 1 µg/kg of remifentanil caused no change in ICP.[239] In a second investigation in craniotomy patients, approximately 0.35 µg/kg/min of
It should be noted that in the aforementioned studies, remifentanil was administered with other drugs that might influence cerebral hemodynamics. More recent studies in human volunteers have demonstrated that the infusion of low (sedative) doses of remifentanil can increase CBF. A PET study in human subjects to whom remifentanil, 0.05 and 0.15 µg/kg/min, was administered revealed increases in CBF in the prefrontal, inferior parietal, and supplementary motor cortices; reductions in CBF were observed in the cerebellum, superior temporal lobe, and midbrain gray matter. [242] The relative increase in CBF was greater with administration of the higher dose of remifentanil. Similar data were obtained by Lorenz and colleagues, who used magnetic resonance imaging for the determination of CBF.[243] [244] The underlying mechanisms for the increases in CBF are not clear. However, the disinhibition produced by low-dose remifentanil infusion may have contributed. In aggregate, the available human data indicate that in low sedative doses, the administration of remifentanil alone can effect minor increases in CBF. With higher doses or with the concomitant administration of anesthetic adjuvants, CBF is either unaltered or modestly reduced.
Benzodiazepines cause parallel reductions in CBF and CMR in humans and monkeys. CBF and CMRO2 decreased by 25% when 15 mg of diazepam was given to head-injured patients.[155] Lorazepam administered to monkeys in doses sufficient to cause sedation reduced CBF by 24% and CMRO2 by 21%.[245] The effects of midazolam on CBF (but not CMR) have also been studied in humans. Forster and associates observed a 30% to 34% reduction in CBF[156] [246] after the administration of 0.15 mg/kg of midazolam to awake healthy human volunteers. Veselis and colleagues, using PET, observed a global reduction in CBF of 12% after a similar dose and noted that the decreases occurred preferentially in the brain regions associated with arousal, attention, and memory.[247] CO2 responsiveness was preserved.[248]
The foregoing studies indicate that benzodiazepines should cause a moderate reduction in CBF in humans and suggest that the effect may be metabolically coupled. The extent of the maximal CBF/CMR reduction produced by benzodiazepines is probably intermediate between the decreases caused by narcotics (modest) and barbiturates (substantial).[21] [249] [250] It appears that benzodiazepines should be safe to administer to patients with intracranial hypertension, provided that respiratory depression and an associated increase in PaCO2 do not occur.
Flumazenil is a highly specific, competitive benzodiazepine receptor antagonist. It had no effect on CBF when administered to unanesthetized human volunteers. [246] [251] In two investigations in dogs with normal intracranial compliance,[249] [252] flumazenil resulted in reversal of the CBF-, CMR-, and ICP-lowering effects of midazolam. Although Knudsen and coworkers[253] observed no change in either CBF or CMR when patients were aroused from midazolam anesthesia with flumazenil at the conclusion of craniotomy for resection of brain tumor, Chiolero and coauthors[254] reported severe increases in ICP when flumazenil was given to midazolam-sedated head-injured patients in whom ICP was poorly controlled before the administration of flumazenil. These latter observations are consistent with two animal investigations. In the investigation of Fleischer and coworkers mentioned earlier,[249] flumazenil not only reversed the CBF and CMR effects of midazolam but also caused a substantial, though short-lived, overshoot above premidazolam levels in both CBF (by 44% to 56%) and ICP (by 180% to 217%). CMR did not rise above control levels, thus indicating that the CBF increase was not metabolically coupled. A similar increase in CBF after flumazenil reversal of midazolam has also been observed in cats.[255] The CBF overshoot effect is unexplained but may be a neurogenically mediated arousal phenomenon.[256] Flumazenil should probably be avoided or used very cautiously to reverse benzodiazepine sedation in patients with impaired intracranial compliance.
No human investigations on the CBF/CMR effects of droperidol in isolation have been conducted. However, the information available from animal investigations and administration of drug combinations in humans[212] [257] [258] [259] [260] taken together suggests that droperidol is not a cerebral vasodilator and probably has little effect on CBF and CMR in humans. The occasional ICP increases that have been observed[258] probably reflect normal autoregulationmediated vasodilation in response to an abrupt fall in MAP.
Among the intravenous anesthetics, ketamine is unique in its ability to cause increases in both CBF and CMR.[157] [261] [262] [263] [264] Animal studies indicate that the changes in CMR are regionally variable. In rats, substantial increases occur in limbic system structures simulatneous with modest changes or small decreases in cortical structures.[262] [265] PET studies in humans have demonstrated that subanesthetic doses of ketamine (0.2 to 0.3 mg/kg) can increase global CMR by about 25%.[266] The greatest increase in CMR occurred in the frontal and anterior cingulate cortex. A relative reduction in CMR in the cerebellum was also observed. Commercially available formulations of ketamine contain both the (S)- and (R)-ketamine enantiomers. (S)-ketamine increases CMR substantially, whereas the (S)-ketamine enantiomer tends to decrease CMR, particularly in the temperomedial cortex and the cerebellum.[267] These changes in CMR are accompanied by corresponding changes in CBF.[268] Because autoregulation is maintained during ketamine anesthesia,[269] the
The anticipated ICP correlate of the increase in CBF has been confirmed to occur in humans.[270] [271] However, anesthetics (diazepam, midazolam, isoflurane/N2 O, propofol) have been shown to blunt or eliminate the ICP or CBF increases associated with ketamine. [264] [271] [272] [273] [274] In fact, decreases in ICP have been reported in response to relatively large doses of ketamine (1.5 to 5 mg/kg) administered to propofol-sedated head-injured patients.[275] Accordingly, although ketamine is probably best avoided as the sole anesthetic in patients with impaired intracranial compliance, it may be reasonable to use it cautiously in patients who are simultaneously receiving the other drugs mentioned earlier.
Lidocaine produces a dose-related reduction in CMRO2 in experimental animals.[276] In dogs, 3 mg/kg lowered CMRO2 by 10%, and 15 mg/kg reduced it by 27%. When very large doses (160 mg/kg) were given to dogs on cardiopulmonary bypass, the reduction in CMRO2 was apparently greater than that seen with high-dose barbiturates. [166] This greater reduction in CMRO2 with lidocaine may occur because the membrane-stabilizing effect of lidocaine also reduces the energy requirement for maintenance of membrane integrity. In unanesthetized human volunteers, Lam and coworkers observed CBF and CMR reductions of 24% and 20%, respectively, after the administration of 5 mg/kg of lidocaine over a 30-minute period followed by an infusion of 45 µg/kg/min.[277]
Bedford and colleagues[278] compared the effectiveness of bolus doses of thiopental (Pentothal), 3 mg/kg, and lidocaine, 1.5 mg/kg, in controlling the acute increase in ICP that occurred after the application of a pinhead holder or skin incision in craniotomy patients. The two regimens were equally effective in causing a reduction in ICP. However, the decrease in MAP was greater with thiopental. Accordingly, a bolus dose of lidocaine is a reasonable adjunct to the prevention or treatment of acute ICP elevation and has been recommended for use in the prevention of increases in ICP associated with endotracheal suctioning. [279] Note that large doses of lidocaine can produce seizures in humans and some experimental animals[280] ; however, lidocaine-induced seizures have not been reported in anesthetized humans. Nonetheless, it seems appropriate to restrict lidocaine doses to amounts that achieve serum levels less than the seizure threshold (>5 to 10 µg/mL) in awake humans. [281] Viegas and Stoelting[282] reported peak serum concentrations of 6.6 to 8.5 µg/mL after a 2-mg/kg bolus of lidocaine. Bolus doses of 1.5 to 2.0 mg/kg therefore seem appropriate.
|
|
|
|
|
|
|
|
|
|
|
|
|