|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Approximately 25% of administered halothane (CF3 CHBrCl) is metabolized to trifluoroacetic acid (CF3 COOH), chloride (Cl- ), and bromide (Br- ) ( Fig. 8-5 ). The major metabolite in humans is trifluoroacetic acid, formed from oxidative metabolism primarily by CYP2E1 and to a lesser extent CYP2A6.[46] The rate-limiting step in oxidative metabolism is breaking of the carbon-hydrogen bond.[47] The first intermediate formed probably is 1,1,1-trifluoro-2-chloro-2-bromoethanol, which would be expected to rapidly decompose to produce hydrogen bromide, and the reactive metabolite, trifluoroacetyl chloride (TFA-Cl). The latter metabolite reacts with water to produce HCl and trifluoroacetic acid, and with phosphatidyl ethanolamine, a membrane phospholipid,[48] to form N-trifluoroacetyl-2-aminoethanol, which has been identified in urine.[49] The TFA-Cl metabolite of halothane also reacts with tissue proteins to form trifluoroacetylated (TFA)-protein adducts (discussed in "Halothane-Associated Immune-Mediated Hepatotoxicity"). Although trifluoroethanol
 Figure 8-5
Major by-products of oxidative and reductive CYP2E1-catalyzed
halothane metabolism.
Figure 8-5
Major by-products of oxidative and reductive CYP2E1-catalyzed
halothane metabolism.
An alternative and minor route of halothane metabolism (<1% of absorbed halothane) is by means of a reductive pathway that requires low oxygen tension and can be catalyzed by CYP2A6 and CYP3A4 (see Fig. 8-5 ).[46] Inorganic Br- and F- are end products of this pathway.[50] [51] [52] Two volatile metabolites (2-chloro-1,1-difluoro-ethylene [CDE] and 2-chloro-1,1,1-trifluorethane [CTE]) and a volatile decomposition product of halothane (2-bromo-2-chloro-1,1-difluoroethylene [DBE]) were first identified in the exhaled gases of patients anesthetized with halothane. [53] The formation of CDE and the release of F- probably result from a CYP-mediated, two-electron reduction of halothane, whereas CTE formation and the production of tissue free radicals result from a CYP-mediated, one-electron reduction.[53] [54] The suicidal inactivation of CYP observed under hypoxic conditions (<40 torr O2 )[55] is presumably the result of the covalent binding of a reactive intermediate of CTE to CYP formed during the metabolism of CTE by CYP. A major pathway of DBE formation is by the base-catalyzed dehydrofluorination of halothane as a result of its interaction with soda lime.[53] [56]
CYP2E1 is inducible by ethanol and by isoniazid and is inhibited by disulfiram.[57] In experimental animals, halothane metabolism is increased after the administration of CYP-inducing agents phenobarbital, [58] Aroclor 1254,[52] and isoniazid.[59] Prolonged exposure to subanesthetic concentrations of halothane results in increased drug metabolism in humans.
Enflurane (CHF2 -O-CF2 -CHCIF) is essentially no longer used in the United States, but examination of its metabolism illustrates how relatively minor changes in chemical structure can dramatically affect the extent of metabolism. Approximately 2.5% of absorbed enflurane is metabolized ( Fig. 8-6 ). Initial oxidation and breaking of the carbonhydrogen bond may occur at the chlorofluoromethyl carbon or at the difluoromethyl carbon. Studies of the metabolism of enflurane with human hepatic microsomes[60] and the isolation of difluoromethoxydifluoroacetic acid from rat liver,[61] human urine,[61] [62] human hepatic microsomes, and cDNA-expressed CYP2E1[60] suggest that primary metabolism occurs at the chlorofluoromethyl carbon. Detection of insignificant amounts of chlorofluoroacetic acid further suggests that there is very little metabolism at the difluoromethyl carbon. The reactive intermediate formed from oxidation at the chlorofluoromethyl carbon can hydrolyze to produce difluoromethoxydifluoroacetic acid or acetylate tissue protein to produce an adduct with immunogenic potential. [63] In either case, inorganic F- is a product of these chemical reactions.
Surgical patients treated on a long-term basis with phenobarbital, phenytoin, or diazepam or who consumed ethanol before anesthesia with enflurane did not have elevated serum F- concentrations compared with untreated patients. In contrast, about 50% of surgical patients on chronic isoniazid therapy demonstrated significantly elevated serum F- concentrations.[12] It is unclear why 50% of patients did not significantly defluorinate enflurane. Studies with purified CYP2E1 from rabbits[64] and humans[65] demonstrate that this CYP isoform is predominantly, if not exclusively, responsible for enflurane defluorination in human liver. Isoniazid treatment seems to significantly enhance enflurane metabolism in vitro in rats,[10] rabbits,[64] and humans.[65] In contrast, treatment of rats with phenobarbital[34] phenytoin,[8] or ethanol[64] [66] increases enflurane defluorination marginally. As with methoxyflurane, defluorination of enflurane in rats decreases after treatment with the CYP inhibitors SKF525A or metyrapone.[34] Continuous exposure of rats to subanesthetic concentrations of enflurane significantly decreases hexobarbital sleeping time, indicating that the metabolism of hexobarbital is induced by enflurane.
Isoflurane (CHF2 -O-CHCl-CF3 ), an isomer of enflurane, is metabolized even more slowly (≅0.2%) than halothane or enflurane (see Fig. 8-6 ). The metabolism of isoflurane results from oxidation of the α-carbon by hepatic CYP2EI.[67] [68] The initial hydroxylated intermediate can decompose to produce a reactive TFA-Cl metabolite identical to halothane or a reactive trifluoroacetyl ester intermediate. Both of these products can be expected to react with water to form trifluoroacetic acid or with protein to produce TFA-protein adducts. Very small amounts of inorganic fluoride are expected to be formed during this metabolism. Although phenobarbital[69] [70] phenytoin,[71] ethanol,[72] and isoniazid[73] [74] pretreatments increase the defluorination of isoflurane, serum F- levels are not clinically significant.[75]
Desflurane (CHF2 -O-CHF-CF3 ) is expected to be metabolized in a manner similar to isoflurane, because these two molecules differ by only one atom at the α-carbon position; desflurane has a fluorine atom, and isoflurane has a chlorine atom (see Fig. 8-6 ). However, the fluorine atom substitution decreases the metabolism at the α-carbon position significantly so that the amount of F- and nonvolatile organic fluorine compounds formed from the metabolism of desflurane is considerably less compared with isoflurane.[76] Peak serum F- concentrations are seen immediately after exposure to desflurane. [77] No increases in serum F- levels above baseline concentrations were measured after volunteers received 7.4 MAC-hours of desflurane. Pretreatment of rats with phenobarbital or ethanol only slightly enhances serum F- concentrations for a brief period.[78]
The rate of sevoflurane [CH2 F-O-CH-(CF3 )2 ] defluorination in vitro is approximately the same as that of methoxyflurane.[79] [80] However, serum F- concentrations after sevoflurane administration are significantly less than those after methoxyflurane, [79] [80] [81] presumably the result of large differences in blood-gas partition coefficients of the two agents (0.69 for sevoflurane versus 10.2 for methoxyflurane). Approximately 5% of absorbed sevoflurane is biotransformed.[81] [82] The fluoromethoxy C-H bond is the site of sevoflurane oxidative metabolism leading to
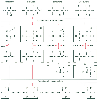 Figure 8-6
Proposed pathways for the CYP2E1-catalyzed metabolism
of halothane, enflurane, isoflurane, and desflurane to acylated hepatic proteins.
For halothane, isoflurane, and desflurane, the trifluoroacetylated protein adducts
are identical in structure, but for enflurane, the adduct is immunologically similar.
Figure 8-6
Proposed pathways for the CYP2E1-catalyzed metabolism
of halothane, enflurane, isoflurane, and desflurane to acylated hepatic proteins.
For halothane, isoflurane, and desflurane, the trifluoroacetylated protein adducts
are identical in structure, but for enflurane, the adduct is immunologically similar.
In a study of human volunteers, hexafluoroisopropanol accounted for 80% of nonvolatile organic fluorine-containing compounds detected in the blood and urine of volunteers anesthetized with sevoflurane.[81] Hexafluoroisopropanol is not subject to further degradation, but it is conjugated to form a glucuronide conjugate.[64] [82] Studies of patients and volunteers have shown that much of the metabolism of sevoflurane to F- occurs during anesthetic exposure, presumably because of the low tissue solubility of sevoflurane and the stability of its metabolites.[85] [86] Peak serum F- concentrations are reached within a few hours of the end of anesthesia. In rats, phenobarbital pretreatment in vivo increased
 Figure 8-7
In vivo metabolism of sevoflurane to inorganic fluoride
(F-
) and hexafluoroisopropanol.
Figure 8-7
In vivo metabolism of sevoflurane to inorganic fluoride
(F-
) and hexafluoroisopropanol.
|
|
|
|
|
|
|
|
|
|
|
|
|