|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ischemic heart disease remains the most common cardiac condition encountered by cardiac anesthesiologists. Despite tremendous advances in our understanding of primary and secondary preventive measures and associated changes in age-adjusted mortality ( Fig. 50-2 ),[14] it is almost
| Structural Change | Functional Change | Ionic, Biophysical/Biochemical Mechanism(s) | Molecular Mechanisms |
|---|---|---|---|
| ↑ Myocyte size | Prolonged contraction | Prolonged cystolic Ca2+ transient |
|
| ↓ Myocyte number |
|
↓ SR Ca2+ pumping rate | ↓ SR Ca2+ |
|
|
|
↓ Pump site density | mRNA |
|
|
|
|
No change in calsequestrin mRNA |
|
|
Prolonged action potential | ↓ ICa inactivation |
|
|
|
|
↓ ITo density |
|
|
|
Diminished contraction velocity | ↓ α-MHC protein | ↓ α-MHC mRNA |
|
|
|
↑ β-MHC protein | ↑ β-MHC mRNA |
|
|
|
↓ Myosin ATPase activity | No change in actin mRNA |
|
|
Diminished β-adrenergic contractile response | ↓ Coupling of BAR-acyclase |
|
|
|
|
↓ TNI phosphorylation |
|
|
|
|
↓ Phospholamban phosphorylation |
|
|
|
|
↓ ICa augmentation |
|
|
|
|
↓ Cai transient augmentation |
|
|
|
|
↑ Enkephalin peptides | ↑ Proenkephalin mRNA |
| ↑ Matrix connective tissue | ↑ Myocardial stiffness | ↑ Atrial natriuretic peptide | ↑ Atrial natriuretic peptide mRNA |
| ↓, Decreased; ↑, increased; BAR, β-adrenergic receptor; SR, sarcoplasmic reticulum. | |||
| From Lakatta EG, Gerstenblith G, Weisfeldt ML: The aging heart: Structure, function and disease. In Braunwald E (ed): Heart Disease: A Textbook of Cardiovascular Medicine. Philadelphia, WB Saunders, 1980, pp 1687–1703. | |||
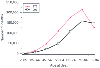 Figure 50-2
Congestive heart disease deaths in the United States.
A comparison of the years 1973 and 1993 reveals that mortality is being postponed
for several years. (Redrawn from Lenfant C: Heart research: Celebration
and renewal. Circulation 96:3822–3823, 1997.)
Figure 50-2
Congestive heart disease deaths in the United States.
A comparison of the years 1973 and 1993 reveals that mortality is being postponed
for several years. (Redrawn from Lenfant C: Heart research: Celebration
and renewal. Circulation 96:3822–3823, 1997.)
Advances in cell and molecular biology over the last 2 to 3 decades have led to a better understanding of the pathogenesis of atherosclerotic disease (including atherosclerotic coronary disease). Atherosclerosis is initiated by the response of the vessel wall (specifically, the endothelium) to injury. Injury to the endothelium can be induced by abnormal flow patterns (e.g., shear stress at points of bifurcation) and is exacerbated by well-established risk factors for coronary artery disease, including hypertension, hyperlipidemia, diabetes, smoking, and likely infections.[15] [16] This theory of the pathogenesis of atherosclerosis has come to be known as "the response to injury" hypothesis.[17] The molecular mechanisms involved are complex, but the interested reader is referred to seminal articles and excellent reviews.[18] [19] [20] [21] [22] Importantly, it is now well established that oxidant signaling under physiologic circumstances, as well as oxidant stress and an imbalance between the production of reactive oxygen species (ROS) and endogenous scavenging mechanisms in pathophysiologic circumstances, are pivotal factors in the development of atherosclerosis in the vasculature. [23] [24] For example, low-density lipoprotein cholesterol requires oxidation before being taken up by cells. The oxidized form that enters cells has potent proinflammatory properties.[25] [26] [27] Oxidized lipid accumulation in cells and the associated inflammatory response leads to foam cell formation and eventual plaque development. Progression of atherosclerosis has been categorized into various phases, as has its correlation with clinical syndromes ( Fig. 50-3 ),[28] [29] including the relationship between plaque stability and acute coronary syndromes (ACSs). These syndromes occur when atherosclerotic plaques rupture or fissure because of degradation of their fibrous caps by enzymes such as metalloproteinases ( Fig. 50-4 ). The release of multiple mediators, including thromboxane, serotonin, and adenosine diphosphate (ADP), causes vasoconstriction,
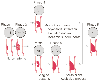 Figure 50-3
Description of lesion morphology and phases of progression
of coronary atherosclerosis in relation to clinical findings. Lipid accumulation,
phases I, II and III; thrombosis and hemorrhage, phases III and IV; and calcification
and fibrous tissue, phases IV and V. Roman numerals indicate lesion types: type
I to III lesions (early lesion) with isolated macrophage/foam cells (I), multiple
foam cell layers (II), or isolated extracellular lipids (III); type IV to Va lesions
(advanced lesions, atheromatous or fibrolipid plaques) with confluent extracellular
lipid pools (atheroma, IV) or fibromuscular tissue layers and atheroma (Va); type
VI lesions (advanced lesions, complicated plaques) with surface defects and/or hemorrhage
and/or thrombi deposition; type Vb to Vc lesions (advanced lesions) with calcifications
(Vb) or fibrous tissue (Vc). (From Fuster V, Gotto AM. Risk reduction.
Circulation 102:IV94–IV102, 2000.)
Figure 50-3
Description of lesion morphology and phases of progression
of coronary atherosclerosis in relation to clinical findings. Lipid accumulation,
phases I, II and III; thrombosis and hemorrhage, phases III and IV; and calcification
and fibrous tissue, phases IV and V. Roman numerals indicate lesion types: type
I to III lesions (early lesion) with isolated macrophage/foam cells (I), multiple
foam cell layers (II), or isolated extracellular lipids (III); type IV to Va lesions
(advanced lesions, atheromatous or fibrolipid plaques) with confluent extracellular
lipid pools (atheroma, IV) or fibromuscular tissue layers and atheroma (Va); type
VI lesions (advanced lesions, complicated plaques) with surface defects and/or hemorrhage
and/or thrombi deposition; type Vb to Vc lesions (advanced lesions) with calcifications
(Vb) or fibrous tissue (Vc). (From Fuster V, Gotto AM. Risk reduction.
Circulation 102:IV94–IV102, 2000.)
The clinical features of ischemic heart disease extend beyond those of angina, myocardial infarction, ischemic cardiomyopathy, and sudden death. They also include the phenomena of stunning, hibernation, and ischemic preconditioning ( Table 50-2 ).[30]
The clinical features of stable angina are well recognized, as are the clinical, electrocardiographic (ECG), and enzymatic features ( Fig. 50-6 ) of ACSs. Such syndromes are now categorized into (1) ST segment elevation myocardial infarction (STEMI) or Q-wave myocardial infarction[18] and (2) non-ST segment elevation ACS (unstable angina), non-ST elevation myocardial infarction (NSTEMI), or non-Q-wave myocardial infarction).[31] [32]
Patients who require surgical revascularization may have presented clinically with stable angina or ACS. The former will have coronary lesions that are not amenable to procedural interventions or will have failed interventional therapy. Patients with ACS usually also present for surgery after interventional thrombolytic therapy. The optimal management (e.g., surgery versus stenting) in specific populations of patients with coronary artery disease is an area of ongoing investigation.[33] Thus, the specific indications for surgical intervention continue to evolve as developments in basic biology and interventional cardiology (e.g., eluting stents) modulate patient management.[34] [35] Medical management of these patients is based on understanding the underlying mechanisms and using target therapies that interrupt these mechanisms. Examples of such therapies include aspirin, heparin, and glycoprotein IIB/IIIA inhibitors.[32] [36] [37] In general, it is appropriate to continue such therapies preoperatively up until full heparinization before CPB because the mechanisms underlying the disease processes are still present. As a corollary, it may also be prudent to not initiate therapies intraoperatively in these patients if such therapies accentuate the mechanisms underlying stable angina and ACS. This is the rationale for not initiating antifibrinolytic therapy until after full heparinization for CPB in these patients.
The need to achieve surgical hemostasis intraoperatively modifies the perioperative management of these patients. For example, most centers discontinue aspirin use 5 to 7 days before cardiac surgery. However, evidence is accumulating that the long-term benefits associated with the continuation of therapies that address the fundamental underlying mechanisms may outweigh the acute perioperative concern of surgical hemostasis.[38] In fact, early use of aspirin in the postoperative phase has also been shown to be associated with a reduced risk of death and ischemic complications.[39]
Coronary ischemia may be unmasked in any patient with ischemic heart disease, be it stable angina or ACS, if myocardial oxygen demand exceeds supply. Therefore, in addition to using therapies directed toward prevention and dissolution of acute thrombus formation on unstable plaque, the physician must manage the patient with a view to optimizing the "external work" of the heart ( Fig. 50-7 ) and the determinants of myocardial oxygen supply and demand ( Fig. 50-8 ). Thus, heart rate and blood pressure control and manipulation of preload and afterload are of paramount importance in successfully managing these patients.
Finally, successful management of these patients is predicated not only on an understanding of the roles of plaque stability and stress-induced infarction and hemodynamics in mediating ischemic events, but also on an appreciation of the interaction between the two ( Fig. 50-9 ). Intuitively we would predict that hemodynamic disturbances have more significant consequences in the setting of tighter lesions ( Fig. 50-9 ). Paradoxically, but not well accepted or understood, myocardial infarctions occur in the setting of mild and moderate coronary lesions (see Fig. 50-9 ). This reflects plaque stability-instability, resulting in acute thrombus formation and the inability of angiography to assess plaque stability.[40] [41] [42]
 Figure 50-4
Mechanisms by which atherosclerotic plaques become ulcerated
(bottom) or fissured (middle).
Top, Inflammation that occurs in selected portions of some atherosclerotic
plaques. Plaque fissuring or ulceration appears to be at least sometimes related
to metalloproteinase release from macrophages present under the thin fibrous cap
(top) or on its surface (middle).
Release of metalloproteinases degrades collagen in the fibrous cap and causes it
to ulcerate or fissure, thereby leading to platelet adhesion, aggregation, and growth
of a thrombus (bottom). Typically, atherosclerotic
plaque that ulcerates has a thin fibrous cap, a large number of inflammatory cells
immediately beneath the cap, and an adjacent lipid pool (top
and bottom). Atherosclerotic plaque that fissures
generally has a thicker cap, a smaller number of inflammatory cells on the surface
of the cap, and a smaller adjacent lipid pool (middle).
LDL, low-density lipoprotein. (Redrawn from Willerson JT, Cohen J [eds]:
Cardiovascular Medicine, 2nd ed. New York, Churchill Livingstone, 2000.)
Figure 50-4
Mechanisms by which atherosclerotic plaques become ulcerated
(bottom) or fissured (middle).
Top, Inflammation that occurs in selected portions of some atherosclerotic
plaques. Plaque fissuring or ulceration appears to be at least sometimes related
to metalloproteinase release from macrophages present under the thin fibrous cap
(top) or on its surface (middle).
Release of metalloproteinases degrades collagen in the fibrous cap and causes it
to ulcerate or fissure, thereby leading to platelet adhesion, aggregation, and growth
of a thrombus (bottom). Typically, atherosclerotic
plaque that ulcerates has a thin fibrous cap, a large number of inflammatory cells
immediately beneath the cap, and an adjacent lipid pool (top
and bottom). Atherosclerotic plaque that fissures
generally has a thicker cap, a smaller number of inflammatory cells on the surface
of the cap, and a smaller adjacent lipid pool (middle).
LDL, low-density lipoprotein. (Redrawn from Willerson JT, Cohen J [eds]:
Cardiovascular Medicine, 2nd ed. New York, Churchill Livingstone, 2000.)
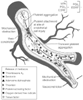 Figure 50-5
Mechanisms involved in thrombosis and vasoconstriction
at sites of atherosclerotic plaque injury resulting from plaque ulceration or fissuring
caused by interventional therapy with angioplasty or stents and other injuries to
endothelium. With such injury, the subendothelium is exposed, platelets adhere and
aggregate, and local accumulation of mediators occurs, largely platelet derived,
including thromboxane A2
, serotonin, adenosine diphosphate, thrombin,
platelet-activating factor, oxygen-derived free radicals, tissue factor, and endothelin.
These mediators promote the growth of thrombus, and most are vasoconstrictors.
Several mediators are also mitogens that promote fibroproliferation or exuberant
scarring after endothelial injury (i.e., "restenotic lesion"). At sites of vascular
injury there are reduced concentrations of normally present endogenous substances
that prevent thrombosis, vasoconstriction, and inflammation, including tissue plasminogen
activator (tPA), prostacyclin (PGI2
), and nitric oxide (tPA, PGI2
,
and endothelium-derived relaxing factor [EDRF] at top right).
Loss of these normally present protective substances helps create a prothrombotic
environment and one in which inflammation and fibroproliferation occur after endothelial
injury. (From Willerson JT, Cohn J [eds]: Cardiovascular Medicine, 2nd
ed. New York, Churchill Livingstone, 2000.)
Figure 50-5
Mechanisms involved in thrombosis and vasoconstriction
at sites of atherosclerotic plaque injury resulting from plaque ulceration or fissuring
caused by interventional therapy with angioplasty or stents and other injuries to
endothelium. With such injury, the subendothelium is exposed, platelets adhere and
aggregate, and local accumulation of mediators occurs, largely platelet derived,
including thromboxane A2
, serotonin, adenosine diphosphate, thrombin,
platelet-activating factor, oxygen-derived free radicals, tissue factor, and endothelin.
These mediators promote the growth of thrombus, and most are vasoconstrictors.
Several mediators are also mitogens that promote fibroproliferation or exuberant
scarring after endothelial injury (i.e., "restenotic lesion"). At sites of vascular
injury there are reduced concentrations of normally present endogenous substances
that prevent thrombosis, vasoconstriction, and inflammation, including tissue plasminogen
activator (tPA), prostacyclin (PGI2
), and nitric oxide (tPA, PGI2
,
and endothelium-derived relaxing factor [EDRF] at top right).
Loss of these normally present protective substances helps create a prothrombotic
environment and one in which inflammation and fibroproliferation occur after endothelial
injury. (From Willerson JT, Cohn J [eds]: Cardiovascular Medicine, 2nd
ed. New York, Churchill Livingstone, 2000.)
Most patients with coronary artery disease who undergo surgical revascularization are well controlled medically. Prudent use of appropriate anesthetic regimens and cardiovascular modulating drugs usually prevents the development of acute ischemia before surgical revascularization is completed. However, the psychological stress associated with surgery, perioperative alterations in medications, and the hemodynamic perturbations associated with anesthetic induction, laryngoscopy, and surgical stimulation may precipitate perioperative ischemia. Such ischemia may be manifested as symptoms (if they occur before induction) or be diagnosed by electrocardiography, echocardiography (regional wall motion abnormalities), or hemodynamics. In this setting, management of ischemia is identical to the management that would be undertaken in a coronary care unit. Pharmacologic manipulation of the determinants of myocardial oxygen supply and demand as outlined in Figure 50-7 and Figure 50-8 is the first step. Pivotal aspects of such manipulation include maintenance of heart rate control (see the section on β-blockers) and adequate coronary perfusion pressure. In addition, despite the caveats discussed in the later section "Nitrates," glyceryl trinitrate (GCN) remains an established therapeutic option for ischemia as long as it does not compromise coronary perfusion pressure. Second, therapies should also be instituted that inhibit thrombus formation on unstable plaque. Such therapies include, at a minimum, heparin and perhaps antiplatelet agents (see the earlier section "Pathophysiology" and the discussion on aspirin). Third, and importantly, intra-aortic balloon pump (IABP) placement should be considered and undertaken if necessary. Indeed, the safety margin provided by placement of an IABP before induction makes this part of the procedure safer. The overall objective should be to stabilize an ischemic patient rather than just hastily proceed to revascularization in an unstable patient with active ischemia.
General cardiology data and studies of the influence of β-blockade on rates of postoperative myocardial infarction suggest that prudent and aggressive heart rate control with β-blockers in patients about to undergo coronary revascularization is appropriate and justified. Tachycardia is an independent predictor of reinfarction in patients who suffer an acute myocardial infarction.[43] [44] Moreover, β-blockers have been demonstrated to improve long-term[45] outcomes in this setting, and this beneficial effect of β-blockade is not confined to patients who have pump failure.[46] [47] In patients undergoing noncardiac surgery, β-blockers have been demonstrated to reduce perioperative ischemia[48] and improve long-term outcomes,[49] and more recently they have also been shown to improve short-term outcomes.[50] Although the mechanisms underlying the beneficial effects of β-blockers may be modestly related to their modulating effect on the predilection for dysrhythmia development or to a reduction in mechanical stress-induced plaque rupture of vulnerable lesions, the primary mechanism underlying their beneficial effects is related to attenuation of tachycardia-induced myocardial ischemia. However, it is now increasingly being recognized that the mechanisms by which β-blockers mediate their effects on coronary blood flow extend beyond that of modulating the major demand variables to also include modulation of supply-side variables such as diastolic interval changes, even at a constant heart rate,[51] changes in endothelial cell function involving endothelin,[52] the redox milieu,[53] and NO.[54] The premise that attenuation of tachycardia-induced ischemia by β-blockers is the primary mechanism of benefit is supported by the observations that (1) the magnitude of the benefits conferred by β-blockade is directly related to the
| Parameter | Conventional Ischemia | Acute Stunning | Chronic Stunning | Hibernation | Preconditioning |
|---|---|---|---|---|---|
| Myocardial function | Reduced | Reduced | Reduced | Reduced | Protected during repeat ischemia by previous ischemia |
| Coronary blood flow | Severely reduced | Postischemic; fully restored | Partially restored | Modestly reduced or possibly normal at rest and repetitively reduced during exercise | Brief ischemia |
|
|
|
|
|
|
→ Fully reperfused |
|
|
|
|
|
|
→ Test for ischemia when reperfused |
| Myocardial energy metabolism | Reduced; increasingly severe as ischemia proceeds | Normal or excessive | Unknown but probably depressed | Reduced in relation to contractile decrease | Reduced ATP demand in test ischemic period |
| Duration | Minutes to hours | Hours to days | Days to weeks to months | Days to months | Protection lasts for hours; may return with "second window" |
| Outcome | Infarction if severe ischemia persists | Full recovery | Incomplete recovery | Recovery if revascularization | Decreased postischemic infarct size; decreased surrogate damage |
| Proposed changes in metabolic regulation of calcium | Insufficient glycolytic ATP to control cell calcium and to prevent reversibility | Cystolic overload and excess oscillations of calcium ions in early reperfusion | Prolonged calcium overload may have led to partial necrosis | Just enough glycolytic ATP to prevent contracture; chronic downregulation and ATP demand | Role of calcium not elucidated, except in hypothesis of repetitive stunning |
| ATP, adenosine triphosphate. | |||||
| From Opie LH: Myocardial reperfusion: New ischemic syndromes. In Opie LH (ed): The Heart, Physiology from Cell to Circulation, 3rd ed. Philadelphia, Lippincott-Raven, 1988, pp 563–588. | |||||
Nitrates were first recommended as treatment of angina by Murrell in 1879.[64] GCN is the organic nitrate available for clinical use and is usually known by the name nitroglycerin. Strictly speaking, the term nitroglycerin is incorrect in that GCN is a nitrate compound (-C-O-NO2 ) and does not contain a nitro group (-C-NO2 ). Nitrates both decrease myocardial oxygen demand and increase myocardial oxygen supply. At the usual clinical doses, nitrates cause vasorelaxation of veins and large conduit arterial vessels. This venodilator-associated decrease in preload is probably the main mechanism underlying relief of angina symptoms after nitrate administration. In ischemic conditions, nitrates exert a favorable influence on myocardial blood flow distribution by increasing the ratio of endocardial to epicardial blood flow. This effect reflects the influence of nitrates on preload and intracavitary pressure as much as the direct effect of nitrates on coronary vessels. Moreover, ischemia is a potent coronary vasodilator, and nitrates are unlikely to exert their main effect via previously maximally dilated resistance vessels. At higher doses, nitrates may also induce vasorelaxation in arteriolar-resistance type vessels. It has been demonstrated that coronary vessels larger than 200 mm are responsive to nitrates whereas vessels smaller than 100 mm exhibit minimal response.[65] This differential response across the vascular tree may reflect in part the local availability of enzymes to convert nitrates to their active metabolites and, ultimately, NO production. The higher oxidative state of nitrates with the requirement for enzyme-mediated
 Figure 50-6
Time course of elevations in serum markers after acute
myocardial infarction (AMI). This figure summarizes the relative timing, rate of
rise, peak values, and duration of elevation above the upper limit of normal for
multiple serum markers after AMI. Although total creatine kinase (CK), CK-MB, and
lactate dehydrogenase (LDH [with isoenzymes]) are traditionally measured, the relatively
slow rate of rise above normal for CK and the potential confusion with noncardiac
sources of enzyme release for both total CK and LDH have inspired the search for
additional serum markers. The smaller molecule myoglobin is released quickly from
infarcted myocardium but is not cardiac specific. Therefore, elevations in myoglobin
that may be detected quite early after the onset of infarction require confirmation
with a more cardiac-specific marker such as CK-MB or troponin I. Troponin I (and
troponin T, not shown) rises more slowly than myoglobin does and may be useful for
diagnosis of infarction even up to 3 to 4 days after the event. Monoclonal antibody
assays for cardiac-specific troponin I and troponin T are now available. (From
Antman EM: General hospital management. In Julian
DG, Braunwald E [eds]: Management of Acute Myocardial Infarction. London, WB Saunders,
1994.)
Figure 50-6
Time course of elevations in serum markers after acute
myocardial infarction (AMI). This figure summarizes the relative timing, rate of
rise, peak values, and duration of elevation above the upper limit of normal for
multiple serum markers after AMI. Although total creatine kinase (CK), CK-MB, and
lactate dehydrogenase (LDH [with isoenzymes]) are traditionally measured, the relatively
slow rate of rise above normal for CK and the potential confusion with noncardiac
sources of enzyme release for both total CK and LDH have inspired the search for
additional serum markers. The smaller molecule myoglobin is released quickly from
infarcted myocardium but is not cardiac specific. Therefore, elevations in myoglobin
that may be detected quite early after the onset of infarction require confirmation
with a more cardiac-specific marker such as CK-MB or troponin I. Troponin I (and
troponin T, not shown) rises more slowly than myoglobin does and may be useful for
diagnosis of infarction even up to 3 to 4 days after the event. Monoclonal antibody
assays for cardiac-specific troponin I and troponin T are now available. (From
Antman EM: General hospital management. In Julian
DG, Braunwald E [eds]: Management of Acute Myocardial Infarction. London, WB Saunders,
1994.)
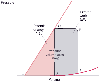 Figure 50-7
Myocardial oxygen consumption correlates with the left
ventricular pressure-volume area (PVA). PVA is in the area of the pressure-volume
(P-V) diagram that is circumscribed by the end-systolic P-V line (E-C), the end-diastolic
P-V relation cure (D-A), and the systolic segment of the P-V trajectory (E-C-D-E).
PVA consists of the external work (EW) performed during systole and the end-systolic
elastic potential energy (PE) stored in the ventricular wall at end-systole. EW
is the area within the P-V loop trajectory (A-B-C-D-A), and PE is the area between
the end-systolic P-V line and the end-diastolic P-V relation curve to the left of
EW (E-C-D-E). (Redrawn from Kameyama T, Asanoi H, Ishizaka S, et al: Energy
conversion efficiency in human left ventricle. Circulation 85:988, 1992.)
Figure 50-7
Myocardial oxygen consumption correlates with the left
ventricular pressure-volume area (PVA). PVA is in the area of the pressure-volume
(P-V) diagram that is circumscribed by the end-systolic P-V line (E-C), the end-diastolic
P-V relation cure (D-A), and the systolic segment of the P-V trajectory (E-C-D-E).
PVA consists of the external work (EW) performed during systole and the end-systolic
elastic potential energy (PE) stored in the ventricular wall at end-systole. EW
is the area within the P-V loop trajectory (A-B-C-D-A), and PE is the area between
the end-systolic P-V line and the end-diastolic P-V relation curve to the left of
EW (E-C-D-E). (Redrawn from Kameyama T, Asanoi H, Ishizaka S, et al: Energy
conversion efficiency in human left ventricle. Circulation 85:988, 1992.)
Mechanistically, nitrate-induced vasorelaxation is mediated by enzymatically induced NO release, activation of guanylyl cyclase in vascular smooth muscle cells, increases in cyclic guanosine monophosphate (cGMP) levels, and modulation of several kinase-dependent enzymes. The end result is a decrease in intracellular calcium concentrations and calcium sensitivity and activation of
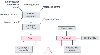 Figure 50-8
Factors influencing myocardial oxygen supply and demand.
(Redrawn from Ardehali A, Ports TA: Myocardial oxygen supply and demand.
Chest 98:699, 1990.)
Figure 50-8
Factors influencing myocardial oxygen supply and demand.
(Redrawn from Ardehali A, Ports TA: Myocardial oxygen supply and demand.
Chest 98:699, 1990.)
 Figure 50-9
Schematic demonstration of the two types of myocardial
infarction (MI). (Redrawn from Landesberg G: The pathophysiology of perioperative
myocardial infarction: Facts and perspectives. J Cardiothorac Vasc Anesth 17:90,
2003.)
Figure 50-9
Schematic demonstration of the two types of myocardial
infarction (MI). (Redrawn from Landesberg G: The pathophysiology of perioperative
myocardial infarction: Facts and perspectives. J Cardiothorac Vasc Anesth 17:90,
2003.)
The issue of nitrate tolerance was initially thought to be of clinical significance only in patients receiving long-acting transdermal therapy. [66] However, it is now clear that tolerance and the pivotal mechanisms underlying tolerance can be induced immediately after nitrate administration.[67] [68] The mechanisms underlying nitrate tolerance are a focus of extensive investigation ( Fig. 50-12 ). It is no longer believed that tolerance is related to limited thiol availability.[69] Moreover, tolerance is most likely not due to impaired nitrate biotransformation. At the intracellular level, tolerance could be due to decreased cGMP
 Figure 50-10
Mechanisms of action of glyceryl trinitrate (GTN). GTN
is metabolized to NO, which stimulates the synthesis of cyclic guanosine monophosphate
(cGMP). In turn, cGMP reduces cytoplasmic Ca2+
by inhibiting inflow and
stimulating mitochondrial uptake, which causes relaxation of smooth muscle cells.
The role of Ca2+
-activated K+
currents is also being investigated;
by inducing hyperpolarization of the cellular membrane, they might contribute to
the limitation of Ca2+
entry into smooth muscle cells, and in endothelial
cells, they might inhibit •
O2
-
production.
By promoting Ca2+
uptake, antithrombin II (AT II) can increase cytoplasmic
Ca2+
and induce Ca2+
-dependent phosphodiesterase 1A1 (PDE1A1).
This may lead to reduced cGMP and cGK activity, thus providing an elegant explanation
for both nitrate tolerance and increased sensitivity to AT III. Some evidence supports
a redox sensitivity of all enzymes involved in these procedures. GTP, guanosine
triphosphate; sGC, soluble guanylyl cyclase; cGK, cGMP-dependent protein kinase.
(Redrawn from Gori T, Parker JD: Nitrate tolerance. A unifying hypothesis.
Circulation 106:2510–2513, 2002.)
Figure 50-10
Mechanisms of action of glyceryl trinitrate (GTN). GTN
is metabolized to NO, which stimulates the synthesis of cyclic guanosine monophosphate
(cGMP). In turn, cGMP reduces cytoplasmic Ca2+
by inhibiting inflow and
stimulating mitochondrial uptake, which causes relaxation of smooth muscle cells.
The role of Ca2+
-activated K+
currents is also being investigated;
by inducing hyperpolarization of the cellular membrane, they might contribute to
the limitation of Ca2+
entry into smooth muscle cells, and in endothelial
cells, they might inhibit •
O2
-
production.
By promoting Ca2+
uptake, antithrombin II (AT II) can increase cytoplasmic
Ca2+
and induce Ca2+
-dependent phosphodiesterase 1A1 (PDE1A1).
This may lead to reduced cGMP and cGK activity, thus providing an elegant explanation
for both nitrate tolerance and increased sensitivity to AT III. Some evidence supports
a redox sensitivity of all enzymes involved in these procedures. GTP, guanosine
triphosphate; sGC, soluble guanylyl cyclase; cGK, cGMP-dependent protein kinase.
(Redrawn from Gori T, Parker JD: Nitrate tolerance. A unifying hypothesis.
Circulation 106:2510–2513, 2002.)
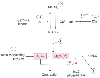 Figure 50-11
Mechanisms of calcium sensitization in vascular smooth
muscle. Sensitization ultimately results from increased functional activity of either
of the contractile proteins actin or myosin. Actin can be modulated by a number
of actin-regulating proteins (1). Myosin activity is determined by its degree of
phosphorylation. Myosin light chain (MLC) phosphorylation can be increased by inhibiting
the mechanisms responsible for dephosphorylation (i.e., inhibiting MLC phosphatase
activity) (2). This can occur either directly or indirectly. It can also be increased
by increasing the activity of MLC kinase. The latter can occur as a result of inhibiting
the breakdown of MLC kinase (3) or as a result of calmodulin activity (4). P, phosphorylation;
MLCKa
, active myosin light chain kinase; MLCKi
, inactive myosin
light chain kinase.
Figure 50-11
Mechanisms of calcium sensitization in vascular smooth
muscle. Sensitization ultimately results from increased functional activity of either
of the contractile proteins actin or myosin. Actin can be modulated by a number
of actin-regulating proteins (1). Myosin activity is determined by its degree of
phosphorylation. Myosin light chain (MLC) phosphorylation can be increased by inhibiting
the mechanisms responsible for dephosphorylation (i.e., inhibiting MLC phosphatase
activity) (2). This can occur either directly or indirectly. It can also be increased
by increasing the activity of MLC kinase. The latter can occur as a result of inhibiting
the breakdown of MLC kinase (3) or as a result of calmodulin activity (4). P, phosphorylation;
MLCKa
, active myosin light chain kinase; MLCKi
, inactive myosin
light chain kinase.
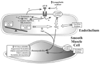 Figure 50-12
Diagram describing the oxidative pathway for the development
of nitrate tolerance. Both endothelial and smooth muscle cells are involved in these
processes. Increased free radical generation, increased production and responsiveness
to angiotensin II (AT II), and L-arginine depletion
may result in nitric oxide synthase (NOS) uncoupling and further production of •
O2
-
and peroxynitrite. BH4
, tetrahydrobiopterin; ET-1, endothelin-1; NADPH,
reduced nicotinamide adenine dinucleotide phosphate; •
O2
-
,
superoxide anion; PKC, protein kinase C. (Redrawn from Gori T, Parker JD:
The puzzle of nitrate tolerance. Pieces smaller than we thought? Circulation 106:2404–2408,
2002.)
Figure 50-12
Diagram describing the oxidative pathway for the development
of nitrate tolerance. Both endothelial and smooth muscle cells are involved in these
processes. Increased free radical generation, increased production and responsiveness
to angiotensin II (AT II), and L-arginine depletion
may result in nitric oxide synthase (NOS) uncoupling and further production of •
O2
-
and peroxynitrite. BH4
, tetrahydrobiopterin; ET-1, endothelin-1; NADPH,
reduced nicotinamide adenine dinucleotide phosphate; •
O2
-
,
superoxide anion; PKC, protein kinase C. (Redrawn from Gori T, Parker JD:
The puzzle of nitrate tolerance. Pieces smaller than we thought? Circulation 106:2404–2408,
2002.)
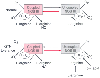 Figure 50-13
Nitric oxide synthase (NOS) dysfunction induced by glyceryl
trinitrate (GTN) therapy. Physiologically, NOS catalyzes a two-step oxidation of
L-arginine to L-citrulline
and NO. Generation of free •
O2
-
is minimal.
The equilibrium between this form of NOS and the uncoupled state (i.e., in which
L-arginine is not oxidized and •
O2
-
is generated) appears to depend on the supply of L-arginine
and the oxidative status of tetrahydrobiopterin (BH4
). During nitrate
tolerance, this equilibrium might be shifted toward the uncoupled form of the enzyme.
In turn, •
O2
-
and peroxynitrite can oxidize
BH4
, thereby resulting in further uncoupling. Folic acid (possibly though
a BH4
-mediated mechanism) and L-arginine
supplementation can reverse this process. BH2
, dihydrobiopterin; NOS
III, endothelial nitric oxide. (Redrawn from Gori T, Parker JD: The puzzle
of nitrate tolerance. Pieces smaller than we thought? Circulation 106:2404–2408,
2002.)
Figure 50-13
Nitric oxide synthase (NOS) dysfunction induced by glyceryl
trinitrate (GTN) therapy. Physiologically, NOS catalyzes a two-step oxidation of
L-arginine to L-citrulline
and NO. Generation of free •
O2
-
is minimal.
The equilibrium between this form of NOS and the uncoupled state (i.e., in which
L-arginine is not oxidized and •
O2
-
is generated) appears to depend on the supply of L-arginine
and the oxidative status of tetrahydrobiopterin (BH4
). During nitrate
tolerance, this equilibrium might be shifted toward the uncoupled form of the enzyme.
In turn, •
O2
-
and peroxynitrite can oxidize
BH4
, thereby resulting in further uncoupling. Folic acid (possibly though
a BH4
-mediated mechanism) and L-arginine
supplementation can reverse this process. BH2
, dihydrobiopterin; NOS
III, endothelial nitric oxide. (Redrawn from Gori T, Parker JD: The puzzle
of nitrate tolerance. Pieces smaller than we thought? Circulation 106:2404–2408,
2002.)
L-Arginine is a critical substrate for NOS and provides the nitrogen for NO production. L-Arginine administration attenuates tolerance to nitrates in both the laboratory[78] and the clinical setting,[79] which suggests that relative lack of available arginine to NOS at the subcellular level may also be partly responsible for tolerance. Finally, the direct central nervous system effects of nitrates (again
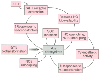 Figure 50-14
Diagram describing the different mechanisms through which
an initial increased production of oxidant free radicals might trigger a series of
autocatalytic processes leading to further •
O2
-
and peroxynitrite generation. Superoxide anion and peroxynitrite may result in uncoupling
of nitric oxide synthase (NOS), sympathetic activation, inhibition of superoxide
dismutase (SOD), and increased production and responsiveness to vasoconstrictors
such as angiotensin II (AT II). These phenomena, in turn, might lead to further
increases in oxidative stress. ET-1, endothelin-1; GTN, glyceryl trinitrate. (Redrawn
from Gori T, Parker JD: Nitrate tolerance. A unifying hypothesis. Circulation
106:2510–2513, 2002.)
Figure 50-14
Diagram describing the different mechanisms through which
an initial increased production of oxidant free radicals might trigger a series of
autocatalytic processes leading to further •
O2
-
and peroxynitrite generation. Superoxide anion and peroxynitrite may result in uncoupling
of nitric oxide synthase (NOS), sympathetic activation, inhibition of superoxide
dismutase (SOD), and increased production and responsiveness to vasoconstrictors
such as angiotensin II (AT II). These phenomena, in turn, might lead to further
increases in oxidative stress. ET-1, endothelin-1; GTN, glyceryl trinitrate. (Redrawn
from Gori T, Parker JD: Nitrate tolerance. A unifying hypothesis. Circulation
106:2510–2513, 2002.)
Heberden first coined the term "angina pectoris" in 1772,[83] even though it was several decades later before it was recognized that angina pectoris had a cardiac origin. Despite occasionally being reported in the literature through the 19th and 20th centuries, the concept that myocardial ischemia can occur without pain did not gain wide acceptance until ambulatory ECG monitoring became available in the 1970s.[84] [85] Moreover, though originally described by Herrick in 1912,[86] the concept that myocardial infarction can also occur without pain took several decades to take hold. It is now recognized that 20% to 60% of myocardial infarctions are unrecognized, and of these, approximately half are truly silent and occur in patients who have no recall of any symptoms.[87] The reason why pain develops in some patients in the setting of myocardial ischemia and not in others is poorly understood. It has been hypothesized either that patients without symptoms have an altered central nervous system response to afferent autonomic signaling [88] or that the pattern of mediator release in ischemic myocardium is altered in a manner that does not achieve a threshold for terminal nerve end stimulation.[89] [90] Silent ischemia is important clinically in that it has at least the same prognostic significance as nonsilent (symptomatic) ischemia.[89] Indeed, it has been demonstrated that patients with nonsilent ischemia may have a preponderance of silent ischemic episodes.[91] The demonstration of myocardial perfusion defects on stress thallium scans and ventricular dysfunction on stress echocardiographic examinations, both in the absence of symptoms, corroborates the clinical significance of silent ischemia,[89] and exercise-induced silent ischemia has been shown to be the most powerful predictor of ischemic heart disease (at least in men).[92]
Syndrome X refers to the phenomenon of anginal chest pain in the setting of angiographically normal coronary vessels and no extracardiac explanation for the symptoms. This phenomenon has been described in up to 20% of patients with chest pain.[93] The mechanisms underlying this syndrome are not well understood, although some evidence supports the concept that coronary microvascular flow may be inadequate or exhibit inadequate reserve if increased blood flow is required.[94] Additionally, such pain could be due to enhanced sensitivity of myocardial pain receptors.[93] The prognosis in these patients is different from that observed in patients with angiographically proven coronary artery disease and, in fact, is similar to the prognosis in the general population.[93]
It is now widely recognized that the response of the myocardium to ischemia is protean. In addition to the conventional manifestations of ischemia, the phenomena of stunning, hibernation, and preconditioning may also represent clinical manifestations of myocardial ischemia (see Table 50-2 ).[95] [96]
Stunning, first described in 1975,[97] refers to the observation that brief periods of ischemia may lead to subsequent myocardial dysfunction that may last for several hours. Importantly, this dysfunction cannot be explained by the development of myocardial necrosis (it is conspicuously absent), nor by ongoing impaired coronary blood flow. Indeed, blood flow during the reperfusion phase of ischemia-reperfusion is essential to the development of stunning. Two important interactive mechanisms underlie stunning.[98] [99] Although the salutary response of the stunned myocardium to inotropes suggests that the availability of intracellular calcium could be impaired, experimental data indicate that myocytes exhibit cytosolic calcium overload. Impaired contraction in the setting of calcium overload indicates decreased sensitivity of the contractile proteins. [100] [101] [102] ROS production after ischemia-reperfusion is an important mechanism in the pathogenesis of stunning. Important types of ROS include superoxide,
Hibernation, first described in 1989,[103] refers to the phenomenon of impaired myocardial function in the setting of ongoing impaired myocardial blood flow. This impaired function is relieved following reinstitution of normal blood flow. Hence, the clinical importance of hibernation is that areas of viable, hibernating myocardium with impaired function are likely to improve after revascularization. Thus, many of the preoperative tests used to evaluate patients with cardiac disease are motivated by this paradigm. The most commonly used tests in this regard are (1) positron emission tomography, the gold standard indicating a mismatch between viable myocardium (assessed with 18 F-labeled deoxyglucose) and myocardial blood flow (assessed with 13 N-labeled ammonia); (2) dobutamine stress tests; and (3) thallium redistribution scans. Noninvasive tests (e.g., contrast-enhanced magnetic resonance imaging) are now being investigated to assess their ability to identify viable myocardium.[104] [105] Areas of myocardium adjacent to an area of infarction frequently exhibit hibernation. Hibernation may also develop in patients without myocardial infarction who have normal resting coronary blood flow but exhibit inadequate blood flow reserve under conditions of increased demand. It has been suggested that these patients may actually represent examples of repetitive stunning.[106] [107] It is clinically important to identify areas of dysfunctional viable myocardium (whether the dysfunction is mediated by hibernation or other mechanisms) because these patients are the ones who will benefit (improvement in symptoms, indices of function and survival) most from revascularization.[108] [109] The mechanisms underlying hibernation are poorly understood but may represent generalized metabolic downregulation and metabolic hypoactivity under conditions of inadequate blood flow. Such a response would represent an adaptive protective process that would be reversed on revascularization. Importantly, it is now increasingly being recognized that the phenomenon of hibernation represents a spectrum of conditions that depend on the severity and duration of impaired blood flow. The latter determine the type and magnitude of local mediator release, which in turn determines the type (irreversible versus reversible) and extent of injury that ensues[30] [110] and whether the injury is reversible. Chronic hibernation may be less readily amenable to reversal after revascularization than the acute or subacute because it also probably involves loss of myofibrils ("disuse atrophy") and reversal to a fetal phenotype with less well differentiated myofibrils, decreased mitochondria, decreased sarcoplasmic reticulum, and other features.[111] [112]
Preconditioning was first described in 1986 by Murray and coworkers. [113] Their canine experiments clearly demonstrated that brief intermittent periods of myocardial ischemia conferred protection against a subsequent larger ischemic insult in that it limited infarct size. These experiments gave rise to the concept of preconditioning. Since then it has clearly been demonstrated that preconditioning is ubiquitous across all species, including humans,[114] and is more pronounced in larger species with lower metabolic rates and slower heart rates. Moreover, preconditioning has clearly been demonstrated in noncardiac tissues.
In the myocardium, the duration of protection after a preconditioning stimulus is limited. Experimental data now indicate that there are two periods, termed windows, of protection after a preconditioning stimulus. The first window of protection occurs within 2 to 3 hours, and a second (late preconditioning) occurs later, between 24 and 96 hours. These two windows have come to be known as the early and late windows of protection or, alternatively, as the classic and delayed windows of protection, respectively. Preconditioning, as originally described, used ischemia as the preconditioning stimulus and limitation in infarct size as the endpoint. However, it is now recognized that several nonpharmacologic (e.g., pacing, exercise) and pharmacologic (e.g., opioids, ROS) stimuli can evoke preconditioning. Moreover, the benefits of preconditioning may extend beyond—and do not necessarily include—limitation of infarct size. For example, preconditioning may confer benefits against the development of dysrhythmia.[115] Thus, discussions of preconditioning must always include a description of the preconditioning stimulus, the specific window of protection in question, and the endpoint being measured to assess efficacy ( Table 50-3 ).
The mechanisms underlying preconditioning, including late preconditioning, are the focus of intense investigations and have been categorized into those that involve triggers, mediators, and effectors ( Fig. 50-15 ).[115] Understanding the mechanisms underlying preconditioning is important because it has the potential to allow one to develop therapeutic strategies to interrogate these mechanisms and pathways and subsequently take advantage of this paradigm in the clinical setting. One mechanism of pivotal importance in preconditioning involves ATP-sensitive potassium channels in the mitochondrial membrane.[116] Activation of these channels with pharmacologic agents (termed pharmacologic preconditioning) has the potential to simulate clinical preconditioning and can be used as a therapeutic modality under controlled circumstances in the clinical setting.
The therapeutic potential of pharmacologic preconditioning requires the ability to predict the ischemic insult. These circumstances occur most frequently during cardiac catheterization and/or cardiac surgery. Although control and predictability of symptoms do not occur in general clinical cardiology, preconditioning may be an important mechanistic and predictive variable. Warm-up angina,
|
|
Protection Against | |||
|---|---|---|---|---|
| Stimulus | Stunning | Arrhythmias | Infarction | Endothelial Dysfunction |
| Nonpharmacologic PC |
|
|
|
|
| Ischemia | + | ? | + | + |
| Heat stress | ? | ? | + | + |
| Rapid pacing | ? | + | ? | ? |
| Exercise | ? | ? | + | ? |
| Pharmacologic PC |
|
|
|
|
| Endotoxin | ? | ? | + | ? |
| Cytokines | ? | ? | + | ? |
| ROS-generating solutions | + | ? | ? | ? |
| NO donors | + | ? | + | ? |
| Adenosine receptor agonists | — | ? | + | ? |
| MLA and analogs | + | ? | + | ? |
| Opioid agonists | ? | ? | + | ? |
| MLA, monophosphoryl lipid A; NO, nitric oxide; PC, preconditioning; ROS, reactive oxygen species. | ||||
| From Gross GJ, Fryer RM: Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res 84:973–979, 1999. By permission of the American Heart Association. | ||||
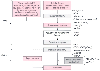 Figure 50-15
Schematic representation of the cellular mechanisms underlying
late preconditioning (PC). A nonlethal cellular stress (e.g., reversible ischemia,
heart stress, ventricular pacing, or exercise) causes release of chemical signals
(nitric oxide [NO], reactive oxygen species [ROS], adenosine, and possibly opioid
receptor agonists) that serve as triggers for the development of late PC. These
substances activate a complex signal transduction cascade that includes protein kinase
C (PKC; specifically, the epsilon isoform), protein tyrosine kinases (PTKs; specifically,
Src and/or Lck), and probably other as-yet-unknown kinases. Similar activation of
PKC and downstream kinases can be elicited pharmacologically by a wide variety of
agents, including naturally occurring—and often noxious—substances (e.g.,
endotoxin, interleukin-1, tumor necrosis factor-α [TNF-α], TNF-β,
leukemia inhibitor factor, or ROS), as well as clinically applicable drugs (NO donors,
adenosine A1
or A3
receptor agonists, endotoxin derivatives,
or δ1
-opioid receptor agonists). Recruitment of PKC and distal
kinases leads to activation of the nuclear factor NF-κB and almost certainly
other transcription factors, and such activation results in increased transcription
and synthesis of multiple cardioprotective proteins that serve as comediators of
protection 2 to 4 days after the PC stimulus. Mediators of late PC identified thus
far include inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), aldose
reductase, and manganese-superoxide dismutase (MnSOD). Among the products of COX-2,
prostaglandin E2
(PGE2
) and prostacyclin (PGI2
)
appear to be the most likely effectors of COX-2-dependent protection. Increased
synthesis of heat shock proteins (HSPs) is unlikely to be a mechanism of late PC,
although the role of post-translational modification of preexisting HSPs remains
to be determined. In addition, the occurrence of cardioprotection on days 2 to 4
requires the activity of PTKs and possibly p38 mitogen-activated protein kinases
(MAPKs), potentially because iNOS and other mediators need to undergo post-translational
modulation to confer protection against ischemia. Opening of adenosine triphosphate-sensitive
potassium (KATP
) channels is also essential for protection against infarction
(but not against stunning). The exact interrelationships among iNOS, COX-2, aldose
reductase, MnSOD, and KATP
channels are unknown, although recent evidence
suggests that COX-2 may be downstream of iNOS (i.e., COX-2 is activated by NO).
Figure 50-15
Schematic representation of the cellular mechanisms underlying
late preconditioning (PC). A nonlethal cellular stress (e.g., reversible ischemia,
heart stress, ventricular pacing, or exercise) causes release of chemical signals
(nitric oxide [NO], reactive oxygen species [ROS], adenosine, and possibly opioid
receptor agonists) that serve as triggers for the development of late PC. These
substances activate a complex signal transduction cascade that includes protein kinase
C (PKC; specifically, the epsilon isoform), protein tyrosine kinases (PTKs; specifically,
Src and/or Lck), and probably other as-yet-unknown kinases. Similar activation of
PKC and downstream kinases can be elicited pharmacologically by a wide variety of
agents, including naturally occurring—and often noxious—substances (e.g.,
endotoxin, interleukin-1, tumor necrosis factor-α [TNF-α], TNF-β,
leukemia inhibitor factor, or ROS), as well as clinically applicable drugs (NO donors,
adenosine A1
or A3
receptor agonists, endotoxin derivatives,
or δ1
-opioid receptor agonists). Recruitment of PKC and distal
kinases leads to activation of the nuclear factor NF-κB and almost certainly
other transcription factors, and such activation results in increased transcription
and synthesis of multiple cardioprotective proteins that serve as comediators of
protection 2 to 4 days after the PC stimulus. Mediators of late PC identified thus
far include inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), aldose
reductase, and manganese-superoxide dismutase (MnSOD). Among the products of COX-2,
prostaglandin E2
(PGE2
) and prostacyclin (PGI2
)
appear to be the most likely effectors of COX-2-dependent protection. Increased
synthesis of heat shock proteins (HSPs) is unlikely to be a mechanism of late PC,
although the role of post-translational modification of preexisting HSPs remains
to be determined. In addition, the occurrence of cardioprotection on days 2 to 4
requires the activity of PTKs and possibly p38 mitogen-activated protein kinases
(MAPKs), potentially because iNOS and other mediators need to undergo post-translational
modulation to confer protection against ischemia. Opening of adenosine triphosphate-sensitive
potassium (KATP
) channels is also essential for protection against infarction
(but not against stunning). The exact interrelationships among iNOS, COX-2, aldose
reductase, MnSOD, and KATP
channels are unknown, although recent evidence
suggests that COX-2 may be downstream of iNOS (i.e., COX-2 is activated by NO).
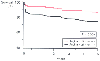 Figure 50-16
Five-year survival curves for patients with [Angina <24
hrs (+)] versus those without [Angina <24 hrs (-)] prodromal angina in the 24
hours before infarction. (Redrawn from Ishihara M, Sato H, Tateishi H, et
al: Implications of prodromal angina pectoris in anterior wall acute myocardial
infarction: Acute angiographic findings and long-term prognosis. J Am Coll Cardiol
30:970–975, 1997.)
Figure 50-16
Five-year survival curves for patients with [Angina <24
hrs (+)] versus those without [Angina <24 hrs (-)] prodromal angina in the 24
hours before infarction. (Redrawn from Ishihara M, Sato H, Tateishi H, et
al: Implications of prodromal angina pectoris in anterior wall acute myocardial
infarction: Acute angiographic findings and long-term prognosis. J Am Coll Cardiol
30:970–975, 1997.)
Anesthetics and their potential interaction with mechanisms underlying preconditioning have emerged as an area of important investigation (also see Chapter 7 ). Moreover, the influence of anesthetics on preconditioning may have important clinical implications.[120] Anesthetics per se do not provoke preconditioning and do not activate the mechanisms underlying preconditioning. However, specific anesthetics in common use (e.g., sevoflurane and isoflurane) have the capacity to profoundly modulate the preconditioning phenomenon when the process itself has been provoked by other triggers.[121] Some anesthetics accentuate the preconditioning response in the presence of triggers, others have no effect and could be termed neutral, and some may have a negative effect by actually attenuating the preconditioning process.[122] Because of the influence of many changing variables, it may be difficult in the clinical setting to determine the impact of specific anesthetics on end points as they relate to preconditioning. However, at least one study has demonstrated the potential salutary effect of volatile anesthetics (specifically sevoflurane) compared with propofol on myocardial function and enzyme leak during cardiac surgery with CPB.[123] [124]
|
|
|
|
|
|
|
|
|
|
|
|
|