|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In developed countries, aortic stenosis is almost always caused by congenital defects in the valve or, in the older population, by the development of "degenerative" (now an outmoded term) calcific aortic stenosis. Aortic stenosis alone is rarely caused by rheumatic disease and, if so, usually occurs with concomitant regurgitation. Congenital valve disease is the single most common congenital heart lesion. Bicuspid aortic valves account for almost all congenital aortic heart lesions and occur in 1% to 2% of the population, mostly males. Unicuspid and tricuspid lesions are rare. Bicuspid aortic valves present at birth generally do not cause clinical problems until the 5th and 6th decades of life. The abnormal valve cusp architecture results in turbulent flow and resultant endothelial damage, which over time causes inflammation and fibrosis. In addition to causing stenosis, bicuspid aortic valves are prone to infection and regurgitation (cusp collapse, fibrotic retraction, secondary to root dilatation). Moreover, they are associated with certain vascular abnormalities, such as medial degeneration, dilatation-aneurysm, and dissection.
Acquired aortic stenosis is a lesion whose incidence is increasing in concert with the aging population. It was previously thought to develop by a "degenerative" process but is now thought to result from an active inflammatory process in a manner not unlike that underlying the pathogenesis of atherosclerotic vascular disease. The early lesions in acquired aortic stenosis are similar to those observed in early atherosclerosis,[125] are exacerbated by some of the same risk factors (e.g., hypercholesterolemia),[126] and may be amenable to similar primary preventive measures such as 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors.[127] Aortic stenosis, whether congenital or acquired, is the most common cardiac valve lesion in the United States.[127]
In aortic stenosis, aortic valve obstruction develops slowly over many years. Left ventricular output is maintained by the development of left ventricular hypertrophy. Parallel replication of sarcomeres results in concentric hypertrophy and is a response to increased intraventricular pressure. Hypertrophy (increased wall thickness) in the presence of increased left ventricular pressure maintains left ventricular wall tension at or near normal values (law of Laplace: Wall tension = [Pressure × Radius] ÷ [Wall thickness × 2]). Left ventricular output can be maintained for many years without ventricular dilatation or symptoms in patients with aortic stenosis. Aortic stenosis is usually considered severe if the systolic gradient exceeds 50 mm Hg or if the effective aortic valve area (AVA) is less then 0.8 cm2 . The hypertrophied ventricle exhibits diminished compliance, as illustrated by an elevated left ventricular end-diastolic pressure (LVEDP) and increased dependence on atrial contraction for ventricular filling. For this reason, prominent "a" waves are seen on left atrial pressure traces in patients with aortic stenosis, and patients experience the precipitous onset of symptoms when they lose their sinus rhythm. As a corollary, maintenance of sinus rhythm is one of the fundamental tenets of managing these patients.
Patients with aortic stenosis are usually asymptomatic and clinically compensated for several decades. The classic symptoms, when they develop, are myocardial ischemia (angina), pulmonary congestion (dyspnea), and inadequate cerebral perfusion (syncope). The pathogenesis of the symptoms that arise in aortic stenosis are illustrated
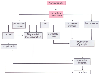 Figure 50-17
Pathophysiology of aortic stenosis. Left ventricular
(LV) outflow obstruction results in increased LV systolic pressure, increased LV
ejection time (LVET), increased LV diastolic pressure, and decreased aortic (Ao)
pressure. Increased LV systolic pressure with LV volume overload increases LV mass,
which may lead to LV dysfunction and failure. Increased LV systolic pressure, LV
mass, and LVET increase myocardial oxygen (O2
) consumption. Increased
LVET results in a decreased myocardial perfusion time. Increased LV diastolic pressure
and decreased Ao diastolic pressure decrease coronary perfusion pressure. Decreased
diastolic time and coronary perfusion pressure decrease myocardial O2
supply. Increased myocardial O2
consumption and decreased myocardial
O2
supply produce myocardial ischemia, which causes further deterioration
in LV function. ↑, Increased; ↓, decreased. (Redrawn from Braunwald
E: Heart Disease: A Textbook of Cardiovascular Medicine. Philadelphia, WB Saunders,
1980, p 1162.)
Figure 50-17
Pathophysiology of aortic stenosis. Left ventricular
(LV) outflow obstruction results in increased LV systolic pressure, increased LV
ejection time (LVET), increased LV diastolic pressure, and decreased aortic (Ao)
pressure. Increased LV systolic pressure with LV volume overload increases LV mass,
which may lead to LV dysfunction and failure. Increased LV systolic pressure, LV
mass, and LVET increase myocardial oxygen (O2
) consumption. Increased
LVET results in a decreased myocardial perfusion time. Increased LV diastolic pressure
and decreased Ao diastolic pressure decrease coronary perfusion pressure. Decreased
diastolic time and coronary perfusion pressure decrease myocardial O2
supply. Increased myocardial O2
consumption and decreased myocardial
O2
supply produce myocardial ischemia, which causes further deterioration
in LV function. ↑, Increased; ↓, decreased. (Redrawn from Braunwald
E: Heart Disease: A Textbook of Cardiovascular Medicine. Philadelphia, WB Saunders,
1980, p 1162.)
Although patients with aortic stenosis may remain asymptomatic for many years, the onset of symptoms has profound prognostic implications. In the absence of surgery, the 5-year survival rate for patients in whom angina develops is 50%. Syncope and dyspnea are associated with a worse prognosis, with 50% alive at 3 and 2 years, respectively.[128] Thus, symptomatic patients with aortic stenosis should undergo aortic valve replacement. Note, however, that the AVA at which symptoms develop is highly variable.[129] Some patients become symptomatic with an AVA of 1 cm2 , whereas others remain asymptomatic with an AVA of less than 0.5 cm2 . Although the Gorlin formula is used to calculate AVA,[127] it has not been well validated for the aortic valve.[130] Furthermore, echoderived Doppler pressure-gradients are sensitive to alterations in transgenic blood flow. Thus, the AVA should be interpreted in the context of all of the patient's data, including symptoms and transaortic velocity gradients (see later).
Two other subgroups of patients, those with asymptomatic severe aortic stenosis and those with severely reduced ejection fractions and aortic stenosis, present more subtle management challenges. The overall incidence of sudden death in asymptomatic patients with severe aortic stenosis is approximately 2%.[128] Although AVA has been used as an indication for surgery in these patients, the threshold value for intervention varies in the range of 0.75 to 1 cm2 . Moreover, in light of the variability in the relationship between symptoms and AVA, there is the potential to operate on most of these patients prematurely, with its attendant morbidity and mortality. Therefore, approaches other than calculations of AVA have been investigated to determine their utility in identifying the subgroup of asymptomatic patients at risk of sudden death. The association of transaortic flow velocity less than 3 m/sec with a low risk and higher than 4 m/sec with a high risk of aortic valve replacement
|
|
True Aortic Stenosis | Pseudo-Aortic Stenosis | ||||
|---|---|---|---|---|---|---|
|
|
Rest | Dobutamine | Nitroprusside | Rest | Dobutamine | Nitroprusside |
| CO, L/min | 3.0 | 5.0 | 3.2 | 3.0 | 5.0 | 4.5 |
| LVP, mm Hg | 130/20 | 160/20 | 120/10 | 130/20 | 140/20 | 120/10 |
| AoP, mm Hg | 90/60 | 100/60 | 80/50 | 90/60 | 100/70 | 90/60 |
| Ğ, mm Hg | 25 | 50 | 30 | 25 | 30 | 25 |
| AVA, cm2 | 0.6 | 0.7 | 0.6 | 0.6 | 0.9 | 0.9 |
| AoP, aortic pressure; AVA, aortic valve area; CO, cardiac output; Ğ, mean gradient, LVP, left ventricular pressure. | ||||||
| Modified from Carabello BA: Evaluation and management of patients with aortic stenosis. Circulation 105:1746–1750, 2002. | ||||||
Patients with severely reduced ejection fractions and aortic stenosis are especially problematic. The operative mortality in a certain subgroup of these patients could reach 21%.[131] In such patients, the reduced ejection fraction can be due to either elevated afterload (mismatch afterload) or irreversible left ventricular dysfunction. The distinction is profoundly important in that the former patients are likely to have a favorable response to aortic valve replacement. Some patients have a condition known as pseudo-aortic stenosis, which is characterized by poor function, mild aortic stenosis, and erroneously calculated AVAs. Distinction between patients with true severe aortic stenosis and those with pseudo-aortic stenosis can be facilitated by measuring transvalvular velocity gradients (a surrogate for afterload mismatch) and comparing the response to inotropes (e.g., dobutamine) and vasodilators on cardiac output, gradient, and calculated AVA ( Table 50-4 ).[128] [131] The administration of pharmacologic agents to aid in this discrimination is not without risk, and thus they should be used only in an aggressively
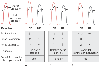 Figure 50-18
The baseline panel shows an example representing a typical
patient with low-gradient aortic stenosis. The hemodynamic response to treatment
with dobutamine (DB) and nitroprusside (NP) helps clarify whether such a patient
has true aortic stenosis (A), relative aortic stenosis
(B), or severe left ventricular dysfunction (C).
A zero denotes no change; a single
arrow, an increase in the value for the particular variable; and two
arrows, a large increase in the value. (Redrawn from Lakatta
E: Aging effects on the vasculature in health: Risk factors for cardiovascular
disease. Am J Geriatr Cardiol 3:11–17, 1994.)
Figure 50-18
The baseline panel shows an example representing a typical
patient with low-gradient aortic stenosis. The hemodynamic response to treatment
with dobutamine (DB) and nitroprusside (NP) helps clarify whether such a patient
has true aortic stenosis (A), relative aortic stenosis
(B), or severe left ventricular dysfunction (C).
A zero denotes no change; a single
arrow, an increase in the value for the particular variable; and two
arrows, a large increase in the value. (Redrawn from Lakatta
E: Aging effects on the vasculature in health: Risk factors for cardiovascular
disease. Am J Geriatr Cardiol 3:11–17, 1994.)
Though originally described over a century ago, the dynamic nature of outflow obstruction was not described until the 1960s.[134] This conspicuous feature of the disease has resulted in not only a proliferation of terms (idiopathic hypertrophic subaortic stenosis, muscular subaortic stenosis) to describe the condition but also a profound underappreciation (at least among noncardiologists) that obstruction occurs in only 25% of these patients.[135] Thus, the term hypertrophic cardiomyopathy (HCM) is the preferred description and applies to patients with ventricular hypertrophy but without an obvious cause such as hypertension or aortic stenosis.
HCM has an incidence of 1 in 500, is inherited as an autosomal dominant lesion, and is the most common genetic cardiac disease. It is also the most common cause of sudden cardiac death (SCD) in children and adolescents. More than 140 mutations on nine different genes coding for sarcomere proteins and two genes coding for cardiac
The clinical manifestations of HCM are heterogeneous, but most patients run a benign course.[138] However, patients with this condition are at risk of SCD and left ventricular outflow obstruction. It is important to identify patients at risk for SCD because implantation of an intracardiac defibrillator is lifesaving. The patient's history, including the presence of syncope, ventricular tachycardia on electrophysiologic studies, a flat or hypotensive response to exercise, and echocardiographic results, has been used to identify those with a higher risk of SCD. A maximum left ventricular wall thickness less than 15 mm is not associated with SCD, but if the left ventricular wall is thicker than 30 mm, SCD occurs at a rate of 2% per year.[139] This finding has been substantiated in a study that also incorporated additional risk factors.[140]
Systolic dynamic obstruction is unique to HCM and occurs in the left ventricular outflow tract between the hypertrophied septum and the anterior leaflet of the mitral valve and the mitral apparatus, which is pulled forward (i.e., toward the septum) during mid to late systole ( Fig. 50-19 ). Several measures (including perioperative interventions) can modulate the propensity for obstruction. Interventions that increase preload, increase intravascular volume, decrease heart rate, decrease inotropy, and increase afterload tend to attenuate obstruction, and vice versa. Obstruction
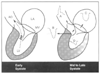 Figure 50-19
Left, Proposed mechanism
of mitral leaflet systolic anterior motion (SAM) in early systole in hypertrophic
cardiomyopathy. Ventricular septal hypertrophy causes a narrowed outflow tract,
as result of which ejection velocity is rapid and the path of ejection (dashed
line) is closer to the mitral leaflets (MV) than normal. This results
in Venturi forces (three short oblique arrows in
the outflow tract) drawing the anterior or posterior mitral leaflets, or both, toward
the septum. Subsequent mitral leaflet-septal contact results in obstruction to left
ventricular (LV) outflow and concomitant mitral regurgitation as seen in the right
panel. By midsystole, SAM-septal contact is well established and is causing marked
narrowing of the LV outflow tract with obstruction to outflow. LA, left atrium;
AO, aorta. Right, Proximal to the level of SAM-septal
contact, converging lines indicate acceleration of
the jet just proximal to the obstruction and narrowing of the jet width. Distal
to the obstruction, the arrow and diverging
lines indicate a high-velocity flow that emanates from the site of SAM-septal
contact and is directed posterolaterally at a considerable angle from the normal
path of aortic outflow. In late systole, although forward flow continues into the
outflow tract and aorta, the volume of flow is much less than in the early nonobstructed
systole. Typical Doppler flow patterns are shown. A, integrated Doppler flow signal
in the ascending aorta; B, high outflow tract velocity recorded by continuous-wave
(CW) Doppler at the site of SAM-septal contact; C, presence of mitral regurgitation
recorded by CW Doppler; D, late systolic velocity peak that can be recorded in the
apical region of the left ventricle. (Redrawn from Wigle ED: Hypertrophic
cardiomyopathy: A 1987 viewpoint. Circulation 75:312, 1987.)
Figure 50-19
Left, Proposed mechanism
of mitral leaflet systolic anterior motion (SAM) in early systole in hypertrophic
cardiomyopathy. Ventricular septal hypertrophy causes a narrowed outflow tract,
as result of which ejection velocity is rapid and the path of ejection (dashed
line) is closer to the mitral leaflets (MV) than normal. This results
in Venturi forces (three short oblique arrows in
the outflow tract) drawing the anterior or posterior mitral leaflets, or both, toward
the septum. Subsequent mitral leaflet-septal contact results in obstruction to left
ventricular (LV) outflow and concomitant mitral regurgitation as seen in the right
panel. By midsystole, SAM-septal contact is well established and is causing marked
narrowing of the LV outflow tract with obstruction to outflow. LA, left atrium;
AO, aorta. Right, Proximal to the level of SAM-septal
contact, converging lines indicate acceleration of
the jet just proximal to the obstruction and narrowing of the jet width. Distal
to the obstruction, the arrow and diverging
lines indicate a high-velocity flow that emanates from the site of SAM-septal
contact and is directed posterolaterally at a considerable angle from the normal
path of aortic outflow. In late systole, although forward flow continues into the
outflow tract and aorta, the volume of flow is much less than in the early nonobstructed
systole. Typical Doppler flow patterns are shown. A, integrated Doppler flow signal
in the ascending aorta; B, high outflow tract velocity recorded by continuous-wave
(CW) Doppler at the site of SAM-septal contact; C, presence of mitral regurgitation
recorded by CW Doppler; D, late systolic velocity peak that can be recorded in the
apical region of the left ventricle. (Redrawn from Wigle ED: Hypertrophic
cardiomyopathy: A 1987 viewpoint. Circulation 75:312, 1987.)
In contrast to systolic obstruction, diastolic dysfunction is observed in most patients with HCM ( Fig. 50-20 ). Moreover, diastolic dysfunction is observed globally in the ventricle, even in areas that are not hypertrophied. Factors that contribute to diastolic dysfunction include increased chamber stiffness secondary to increased muscle mass, decreased ventricular volume and increased intrinsic muscle stiffness, decreased relaxation secondary to decreased relaxation times, and nonuniformity of the disease. The diastolic dysfunction most likely contributes to increased left atrial and pulmonary pressures and the clinical symptoms of dyspnea. Myocardial ischemia can occur in patients with HCM for reasons similar to those found in aortic stenosis.
Medical management of patients with HCM is predicated on maintenance of euvolemia to hypervolemia; use of negative inotropes such as β-blockers, calcium antagonists, and disopyramide; and maintenance of afterload. In addition to placement of an intracardiac defibrillator for protection against SCD, DDD pacing and alcohol ablation[135] of the septum are other nonoperative treatment options. Myotomy-myomectomy is the most frequently used surgical approach. However, mitral valve replacement has also been used with good results because it completely relieves systolic anterior motion of the anterior leaflet of the mitral valve and its contribution to obstruction. It may be the preferred option in patients who have failed myotomy-myomectomy, in patients with less than 18-mm thickness of the upper septum, and in those with intrinsic mitral valve disease. [141]
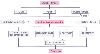 Figure 50-20
Diastolic dysfunction in hypertrophic cardiomyopathy
(HCM). Increased chamber stiffness or decreased compliance occurs as a result of
increased muscle mass and the resulting decreased ventricular volume. Increased
muscle stiffness also occurs as a result of myocardial fibrosis. Thus, all three
factors that affect the stiffness or compliance of the ventricle are altered in a
way that increases chamber stiffness. Left ventricular relaxation in HCM is impaired
because of changes in loading conditions, decreased inactivation, and increased nonuniformity.
The subaortic stenosis in obstructive HCM represents a contraction load on the ventricle
that delays and impairs relaxation. Coronary and ventricular filling loads, which
aid in relaxation, are reduced in HCM because of the degree of hypertrophy and other
reasons. High myoplasmic calcium results in decreased inactivation, which impairs
relaxation both directly and indirectly by the relaxation process. Finally, much
nonuniformity exists in HCM, another factor that also impairs relaxation. Thus,
all three factors controlling relaxation are altered to impair it in HCM. (Redrawn
from Wigle ED, et al: Hypertrophic cardiomyopathy. In
Abelmann WH, Braunwald E [eds]: Cardiomyopathies, Myocarditis, and Pericardial Disease.
Atlas of Heart Diseases, vol 2. Philadelphia, Current Medicine, 1995.)
Figure 50-20
Diastolic dysfunction in hypertrophic cardiomyopathy
(HCM). Increased chamber stiffness or decreased compliance occurs as a result of
increased muscle mass and the resulting decreased ventricular volume. Increased
muscle stiffness also occurs as a result of myocardial fibrosis. Thus, all three
factors that affect the stiffness or compliance of the ventricle are altered in a
way that increases chamber stiffness. Left ventricular relaxation in HCM is impaired
because of changes in loading conditions, decreased inactivation, and increased nonuniformity.
The subaortic stenosis in obstructive HCM represents a contraction load on the ventricle
that delays and impairs relaxation. Coronary and ventricular filling loads, which
aid in relaxation, are reduced in HCM because of the degree of hypertrophy and other
reasons. High myoplasmic calcium results in decreased inactivation, which impairs
relaxation both directly and indirectly by the relaxation process. Finally, much
nonuniformity exists in HCM, another factor that also impairs relaxation. Thus,
all three factors controlling relaxation are altered to impair it in HCM. (Redrawn
from Wigle ED, et al: Hypertrophic cardiomyopathy. In
Abelmann WH, Braunwald E [eds]: Cardiomyopathies, Myocarditis, and Pericardial Disease.
Atlas of Heart Diseases, vol 2. Philadelphia, Current Medicine, 1995.)
|
|
|
|
|
|
|
|
|
|
|
|
|