 |
|
|
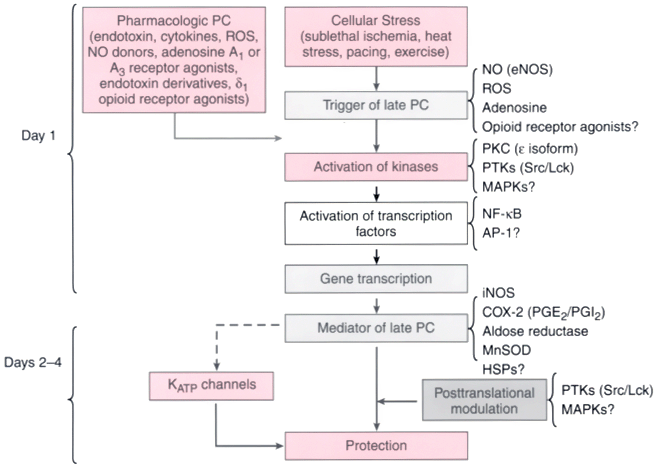
Figure 50-15
Schematic representation of the cellular mechanisms underlying
late preconditioning (PC). A nonlethal cellular stress (e.g., reversible ischemia,
heart stress, ventricular pacing, or exercise) causes release of chemical signals
(nitric oxide [NO], reactive oxygen species [ROS], adenosine, and possibly opioid
receptor agonists) that serve as triggers for the development of late PC. These
substances activate a complex signal transduction cascade that includes protein kinase
C (PKC; specifically, the epsilon isoform), protein tyrosine kinases (PTKs; specifically,
Src and/or Lck), and probably other as-yet-unknown kinases. Similar activation of
PKC and downstream kinases can be elicited pharmacologically by a wide variety of
agents, including naturally occurring—and often noxious—substances (e.g.,
endotoxin, interleukin-1, tumor necrosis factor-α [TNF-α], TNF-β,
leukemia inhibitor factor, or ROS), as well as clinically applicable drugs (NO donors,
adenosine A1
or A3
receptor agonists, endotoxin derivatives,
or δ1
-opioid receptor agonists). Recruitment of PKC and distal
kinases leads to activation of the nuclear factor NF-κB and almost certainly
other transcription factors, and such activation results in increased transcription
and synthesis of multiple cardioprotective proteins that serve as comediators of
protection 2 to 4 days after the PC stimulus. Mediators of late PC identified thus
far include inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), aldose
reductase, and manganese-superoxide dismutase (MnSOD). Among the products of COX-2,
prostaglandin E2
(PGE2
) and prostacyclin (PGI2
)
appear to be the most likely effectors of COX-2-dependent protection. Increased
synthesis of heat shock proteins (HSPs) is unlikely to be a mechanism of late PC,
although the role of post-translational modification of preexisting HSPs remains
to be determined. In addition, the occurrence of cardioprotection on days 2 to 4
requires the activity of PTKs and possibly p38 mitogen-activated protein kinases
(MAPKs), potentially because iNOS and other mediators need to undergo post-translational
modulation to confer protection against ischemia. Opening of adenosine triphosphate-sensitive
potassium (KATP
) channels is also essential for protection against infarction
(but not against stunning). The exact interrelationships among iNOS, COX-2, aldose
reductase, MnSOD, and KATP
channels are unknown, although recent evidence
suggests that COX-2 may be downstream of iNOS (i.e., COX-2 is activated by NO).
|
|