|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The role of the kidney in controlling the interior milieu is modulated by a complex set of interactions. Two mutually dependent, but opposing neurohormonal systems maintain blood pressure, intravascular volume, and salt and water homeostasis ( Fig. 20-13 ). The sympathoadrenal axis, the renin-angiotensin-aldosterone system, and AVP defend against hypotension and hypovolemia by promoting vasoconstriction and salt and water retention. The prostaglandins, bradykinins, and ANPs defend against hypertension and hypervolemia by promoting vasodilation and salt and water excretion ( Fig. 20-14 ).
Anesthesia does not perturb these systems to any substantial degree. A considerable body of work suggests that in an intact organism, anesthetics affect renal function through extrarenal circulatory changes rather than by direct actions on the kidney.[30] Surgical or traumatic injury, on the other hand, induces profound vasoconstriction and salt and water retention, which may persist for several days. The clinical sequela is postoperative oliguria and edema. Renal vasoconstriction also predisposes the kidney to further perioperative ischemic and nephrotoxic insults. Elucidation of the role of ANP enhances the concept that these changes can be prevented or modified by maintenance of normal or increased intravascular (and thereby atrial) volume.
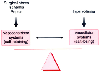 Figure 20-13
Neurohormonal regulation of renal function. Normally,
a balance is present between systems promoting renal vasoconstriction and sodium
retention versus systems promoting renal vasodilation and sodium excretion. Surgical
stress, ischemia, and sepsis tip the balance in favor of vasoconstriction and sodium
retention. On the other hand, hypervolemia (or induction of atrial stretch) tips
the balance in favor of vasodilation and sodium excretion.
Figure 20-13
Neurohormonal regulation of renal function. Normally,
a balance is present between systems promoting renal vasoconstriction and sodium
retention versus systems promoting renal vasodilation and sodium excretion. Surgical
stress, ischemia, and sepsis tip the balance in favor of vasoconstriction and sodium
retention. On the other hand, hypervolemia (or induction of atrial stretch) tips
the balance in favor of vasodilation and sodium excretion.
Sympathetic effects on the kidney are mediated by circulating epinephrine and neuronal release of norepinephrine. The renal cortex has a dense plexus of autonomic nerve fibers derived from the T12 to L4 spinal segments by way of the celiac plexus. The primary stimulus to the sympathetic response is a decrease in arterial blood pressure sensed by baroreceptors in the aortic arch, carotid sinus, and afferent arteriole. Afferent fibers travel by means of the vagus nerve and decrease the impulse transmission rate to the mediating centers in the hypothalamus, which results in increased adrenergic nerve activity. The kidney does not have any parasympathetic innervation.
A G protein-coupled phospholipase C receptor populates vascular smooth muscle and the mesangium and responds to α-adrenergic stimulation by epinephrine
 Figure 20-14
Neurohormonal renal regulatory systems. GFR, glomerular
filtration rate; RBF, renal blood flow. (Modified from Sladen RN: Effect
of anesthesia and surgery on renal function. Crit Care Clin 3:380, 1987. ©
1987, The Williams and Wilkins Company, Baltimore.)
Figure 20-14
Neurohormonal renal regulatory systems. GFR, glomerular
filtration rate; RBF, renal blood flow. (Modified from Sladen RN: Effect
of anesthesia and surgery on renal function. Crit Care Clin 3:380, 1987. ©
1987, The Williams and Wilkins Company, Baltimore.)
Mild α-adrenergic stimulation appears to cause preferential efferent arteriolar constriction, which preserves the FF. Severe α-adrenergic stimulation causes predominant afferent arteriolar constriction and decreases the FF.[31] Thus, the adrenergic response to a moderate decrease in renal perfusion (e.g., general anesthesia) favors preservation of the GFR. In contrast, the adrenergic response to shock exacerbates the decrease in GFR already induced by renal hypoperfusion (see Fig. 20-4 ).
Adrenergic nerves also supply the proximal tubule, thick ascending limb of Henle, and collecting duct, and their stimulation enhances NaCl reabsorption at these sites. Gas tracer studies suggested that sympathetic activation causes sodium retention by intrarenal redistribution of RBF from the outer cortex to salt-retaining juxtamedullary nephrons, but this effect was not confirmed by microsphere studies. [28] [29]
A close relationship has been found between sympathetic stimulation and activation of the renin-angiotensin system. Adrenergic stimulation releases renin from the juxtaglomerular apparatus, and adrenergically induced vasoconstriction can be blocked by angiotensin-converting enzyme (ACE) inhibitor drugs such as captopril.
The effects of the administration of exogenous adrenergic agonists depend on their agonist activity. Drugs with predominantly α-adrenergic effects, such as norepinephrine, epinephrine, phenylephrine, and high-dose dopamine (>10 µg/kg/min), exacerbate the endogenous sympathetic responses to hypotension (see also Chapter 16 ). Drugs with predominantly β1 - and β2 -adrenergic activity, such as dobutamine or isoproterenol, cause marked increases in cardiac output and thus RBF, but it is difficult to ascertain their intrarenal effects. Dopaminergic agonists, such as low-dose dopamine (<3 µg/kg/min), dopexamine, and fenoldopam, selectively increase RBF and may oppose α-adrenergic renal vasoconstriction. [24] They induce a quite different response in the kidney and will be discussed separately.
The juxtaglomerular apparatus consists of three groups of specialized tissue. In the afferent arteriole, modified fenestrated endothelial cells produce renin; in the juxtaposed distal tubule, cells of the macula densa act as chemoreceptors; and in the glomerulus, mesangial cells have contractile properties (see Fig. 20-3 ). Together, these cells provide an important regulating system for blood pressure, salt, and water homeostasis.[32]
Renin secretion is stimulated by actual hypovolemia (hemorrhage, diuresis, sodium loss or restriction) or effective hypovolemia (positive-pressure ventilation, congestive heart failure, sepsis, or cirrhosis with ascites). Its release is controlled by several mechanisms. A decrease in renal artery perfusion pressure triggers baroreceptors in the afferent arterioles. Sympathetic nerve stimulation and circulating catecholamines act on β-adrenergic receptors in the afferent arterioles. An increase in chloride concentration in the distal tubular fluid activates cells of the macula densa, which trigger renin release from the afferent arteriole. This tubuloglomerular feedback appears to play a role in modulating GFR during normal and abnormal renal function through a continuous feedback loop.[4] [22]
Renin acts on angiotensinogen, a large circulating glycoprotein released from the liver, and cleaves off a decapeptide, angiotensin I. In the kidney and the lung, angiotensin I is further cleaved by endothelial-based ACE to form an octapeptide, angiotensin II, a potent vasoconstrictor ( Fig. 20-15 ). Renin is the rate-limiting enzyme in the production of angiotensin II.[33]
Activation of modest amounts of angiotensin II causes renal cortical vasoconstriction predominantly at the level of the efferent arterioles (see Fig 20-4 ). This vasoconstriction acts to maintain the glomerular FF in the face of mild to moderate decreases in RBF or perfusion pressure. The importance of this protective mechanism is emphasized by the deterioration in GFR that occurs when ACE inhibitors are administered to patients with hypotension, renal insufficiency, or unilateral renal artery stenosis.[34] [35] Severe stress induces the release of high levels of angiotensin II, which constricts the glomerular mesangial cells and decreases the glomerular FF. Angiotensin II promotes systemic vasoconstriction at about one tenth of its renal effect, yet systemic arterial pressure and vascular resistance can be markedly decreased by ACE inhibitors such as captopril and enalapril or angiotensin receptor antagonists.
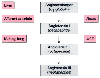 Figure 20-15
Renin-angiotensin system. For an explanation, see the
text. ACE, angiotensin-converting enzyme.
Figure 20-15
Renin-angiotensin system. For an explanation, see the
text. ACE, angiotensin-converting enzyme.
Angiotensin II triggers a number of responses that modulate or oppose its own actions. It inhibits renin secretion by a negative feedback mechanism. Blockade of angiotensin formation by ACE inhibitors causes vasodilation but increases plasma renin levels. Angiotensin II activates phospholipase A2 , which triggers the synthesis of intrarenal prostaglandins. Vasodilator prostaglandins modulate the action of angiotensin II and may be responsible for its preferential activity on the efferent arteriole at low plasma levels.[33] Angiotensin-induced vasoconstriction increases atrial pressure and releases ANP, which opposes the renin-angiotensin-aldosterone system.
The consequences of ACE inhibition on renal function depend on the patient's volume status, systemic hemodynamics, and baseline renal perfusion. In the long-term treatment of hypertension and congestive heart failure, especially in diabetics, the administration of ACE inhibitors such as captopril, enalapril, or lisinopril decreases renal vascular resistance and appears to benefit renal function. Short-term pretreatment with captopril may prevent a decrease in RBF and GFR and preserve sodium excretion during CPB.[36] However, deterioration in renal function and hyperkalemia have been reported with the use of ACE inhibitors in patients with hypotension, renal insufficiency, or unilateral renal artery stenosis, probably related to the blockade of compensatory angiotensin-mediated efferent arteriolar constriction.[35] It may be prudent to avoid their use in patients with unstable hemodynamics in the immediate perioperative period.
Aldosterone is a steroid hormone secreted by the zona glomerulosa of the adrenal cortex in response to hyperkalemia or hyponatremia. Angiotensin II and adrenocorticotropic hormone also trigger its release. It acts at the thick ascending limb of the loop of Henle, the principal cells of the distal tubule, and the collecting duct to increase active absorption of sodium and passive absorption of water, thus culminating in an expanded blood volume. Sodium retention in vessel walls appears to enhance their response to vasoconstrictor agents.
In contrast to the immediate sympathetic angiotensin II response to hypovolemia, there is a delay of about 1 to 2 hours from the secretion of aldosterone to its action on sodium reabsorption. As illustrated in Figure 20-16 , aldosterone forms a complex with a receptor at the cell membrane in the principal cells of the distal tubule. The aldosterone-receptor complex travels to the cell nucleus, where it induces cytoplasmic transcription of mRNA. Such transcription fosters the synthesis of proteins that form sodium channels in the apical cell membrane and thus enhances the Na-K-ATPase pump in the basolateral cell membrane.[37] Sodium is transported from the tubular fluid into the peritubular capillary in exchange for potassium. Long-standing stimulation of aldosterone secretion, characteristically induced by the intravascular
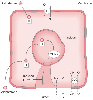 Figure 20-16
Action of aldosterone. Aldosterone enters the distal
tubular cytoplasm, attaches to a receptor, and then migrates to the nucleus, where
it induces the formation of messenger RNA (mRNA). The mRNA in turn induces the synthesis
of a protein that enhances the permeability of the apical (luminal) membrane to sodium
and potassium. Reabsorption of sodium stimulates the basolateral membrane Na-K-ATPase
pump, the intracellular concentration of potassium rises, and it follows its concentration
gradient out into the lumen. The net effect of aldosterone's action is sodium reabsorption
and potassium loss. CO, cotransporter (= symporter); P, sodium-potassium-ATPase
pump; R, receptor. (Redrawn from Wingard LB, Brody TM, Larner J, Schwartz
A: Diuretics: Drugs that increase excretion of water and electrolytes. In
Wingard LB, Brody TM, Larner J, Schwartz A [eds]: Human Pharmacology: Molecular-to-Clinical.
London, Wolfe, 1991, p 249.)
Figure 20-16
Action of aldosterone. Aldosterone enters the distal
tubular cytoplasm, attaches to a receptor, and then migrates to the nucleus, where
it induces the formation of messenger RNA (mRNA). The mRNA in turn induces the synthesis
of a protein that enhances the permeability of the apical (luminal) membrane to sodium
and potassium. Reabsorption of sodium stimulates the basolateral membrane Na-K-ATPase
pump, the intracellular concentration of potassium rises, and it follows its concentration
gradient out into the lumen. The net effect of aldosterone's action is sodium reabsorption
and potassium loss. CO, cotransporter (= symporter); P, sodium-potassium-ATPase
pump; R, receptor. (Redrawn from Wingard LB, Brody TM, Larner J, Schwartz
A: Diuretics: Drugs that increase excretion of water and electrolytes. In
Wingard LB, Brody TM, Larner J, Schwartz A [eds]: Human Pharmacology: Molecular-to-Clinical.
London, Wolfe, 1991, p 249.)
AVP, previously known as antidiuretic hormone, regulates urinary volume and osmolality and controls diuresis and antidiuresis. It is a nine-amino acid peptide, 8-arginine-vasopressin, that is synthesized in the supraoptic and paraventricular nuclei of the anterior hypothalamus.[37] These nuclei are essentially the cell bodies of neurons whose axons extend down into nerve terminals in the posterior pituitary, together constituting the neurohypophysis. When AVP is synthesized, it under-goes neuroaxonal transport to the posterior pituitary gland, where it is stored in granules. Neural stimulation of the cell bodies triggers exocytosis of AVP from the terminal vesicles into the circulation.
AVP acts on specific V2 receptors in the collecting ducts to induce water reabsorption and a decreased flow of concentrated urine. It also increases NaCl reabsorption from
The V2 receptor on the basolateral cell membrane of the collecting duct responds to AVP through a receptor mechanism analogous to the β-adrenergic receptor.[37] By activation of G protein-coupled adenylyl cyclase, ATP is converted to cyclic adenosine monophosphate (cAMP). In turn, cAMP activates a protein kinase that causes preformed vesicles containing water channels to migrate and fuse with the apical cell membrane. This action results in a dramatic increase in membrane permeability to water, which is reabsorbed into the cell and thence into the peritubular capillary. This process is rapidly reversed when plasma AVP levels decline (the plasma half-life of AVP is between 5 and 15 minutes).
Hypothalamic osmoreceptors are sensitive to increases in serum osmolality of as little as 1% above normal. As illustrated in Figure 20-17A , the threshold for AVP secretion (and the sensation of thirst) is between 280 and 290 mOsm/kg. Once this threshold is exceeded, the secretion rate has a very steep gain.[38] Even mild dehydration results in rapid antidiuresis, and urine osmolality can increase from 300 to 1200 mOsm/kg as plasma AVP levels rise from 0 to 5 pg/mL (see Fig. 20-17B ).
Decreases in intravascular volume also stimulate AVP secretion mediated by stretch receptors with vagal afferents in the left atrium and pulmonary veins. Hypovolemia-induced secretion of AVP overrides the osmolar responses and contributes to the perioperative syndrome of inappropriate antidiuretic hormone secretion (SIADH): fluid retention, hypo-osmolality, and hyponatremia.[39] The situation is exacerbated by the administration of large volumes of hypotonic solutions that decrease serum osmolality. Psychic stress, through cortical input, also induces AVP release and can override osmotic and volume sensors.
By far the most potent trigger for AVP release is systemic arterial hypotension mediated by aortic and carotid baroreceptors. It overrides all other triggers, and plasma AVP may reach levels 10- to a 1000-fold greater than normal (see Fig. 20-17C ). At these concentrations AVP acts as a vasoconstrictor, especially in the outer renal cortex. It does so by stimulating the V1 receptor, which is found on vascular smooth muscle cells, glomerular mesangial cells, and cells of the vasa recta and promotes vasoconstriction through the phosphatidylinositol pathway.[38] AVP maintains effective glomerular filtration pressure because it is an extremely potent constrictor of the efferent arteriole, and unlike catecholamines and angiotensin, it has little effect on the afferent arteriole, even at high plasma levels.[40]
Anesthetic agents have little direct effect on AVP secretion, except through the changes that they induce in
 Figure 20-17
Physiologic regulation of arginine vasopressin (AVP).
For an explanation, see the text. (From Landry DW: Vasopressin deficiency
and hypersensitivity in vasodilatory shock: Discovery of a new clinical syndrome.
P & S Med Rev 3:3–7, 1996.)
Figure 20-17
Physiologic regulation of arginine vasopressin (AVP).
For an explanation, see the text. (From Landry DW: Vasopressin deficiency
and hypersensitivity in vasodilatory shock: Discovery of a new clinical syndrome.
P & S Med Rev 3:3–7, 1996.)
|
|
|
|
|
|
|
|
|
|
|
|
|