|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Intrarenal prostaglandins play an important role in endogenous renal protection, largely by vasodilating juxtamedullary blood vessels and maintaining inner
Phospholipase A2 , which resides in the inner lipid layer of the cell membrane, controls prostaglandin production through its formation of the prime precursor arachidonic acid. It is stimulated by ischemia and hypotension and also by norepinephrine, angiotensin II, and AVP. Thus, the factors that induce and mediate the stress response simultaneously activate prostaglandins, which defend the kidney against their actions. Cyclooxygenase-1 (COX-1) acts on arachidonic acid to form PGG2 , the precursor of the family of vasodilator prostaglandins that includes PGD2 , PGE2 , and PGI2 (prostacyclin). They induce vasodilation through activation of cAMP, which blocks distal tubule sodium reabsorption, and they oppose the actions of norepinephrine, angiotensin II, and AVP. Prostaglandins may be particularly important in decreasing the vasoconstrictor activity of angiotensin II on the afferent arteriole and glomerular mesangial cells. [33] Production of prostaglandins promotes renal vasodilation, maintains intrarenal hemodynamics, and enhances sodium and water excretion. The renal vasodilator response to mannitol during hypoperfusion appears to be mediated through prostaglandin activation.[42] At the same time, prostaglandins also stimulate renin secretion, so a constant "yin and yang" occurs between the two systems.[43]
COX-2 forms derivatives of arachidonic acid that induce inflammation and renal vasoconstriction, and these derivatives are thus important in pathologic states. Thromboxane A2 is derived from cyclic endoperoxides by
 Figure 20-18
Synthesis of renal prostaglandins. Phospholipase A2
is stimulated by ischemia, norepinephrine, and angiotensin II and cleaves arachidonic
acid from its bond with membrane phospholipid. Cyclooxygenase acts on arachidonic
acid to form evanescent cyclic endoperoxides (PGG2
and PGH2
).
The action of isomerase and prostacyclin synthetase culminates in formation of the
vasodilator prostaglandins PGD2
, PGE2
, and PGI2
(prostacyclin), which oppose the action of the renin-angiotensin system on the kidney
and protect against ischemic stress. Inhibition of cyclooxygenase by nonsteroidal
anti-inflammatory drugs predisposes the kidney to damage. Under hypoxic or ischemic
conditions, cyclic endoperoxides undergo reduction to the vasoconstrictor PGF2
,
which acts on thromboxane receptors. Endotoxin increases the activity of leukocyte
lipoxygenase and thromboxane synthetase. Leukotrienes (especially C4
and D4
) and thromboxane (TXA2
) induce renal vasoconstriction
and contribute to the vasomotor nephropathy of sepsis.
Figure 20-18
Synthesis of renal prostaglandins. Phospholipase A2
is stimulated by ischemia, norepinephrine, and angiotensin II and cleaves arachidonic
acid from its bond with membrane phospholipid. Cyclooxygenase acts on arachidonic
acid to form evanescent cyclic endoperoxides (PGG2
and PGH2
).
The action of isomerase and prostacyclin synthetase culminates in formation of the
vasodilator prostaglandins PGD2
, PGE2
, and PGI2
(prostacyclin), which oppose the action of the renin-angiotensin system on the kidney
and protect against ischemic stress. Inhibition of cyclooxygenase by nonsteroidal
anti-inflammatory drugs predisposes the kidney to damage. Under hypoxic or ischemic
conditions, cyclic endoperoxides undergo reduction to the vasoconstrictor PGF2
,
which acts on thromboxane receptors. Endotoxin increases the activity of leukocyte
lipoxygenase and thromboxane synthetase. Leukotrienes (especially C4
and D4
) and thromboxane (TXA2
) induce renal vasoconstriction
and contribute to the vasomotor nephropathy of sepsis.
Kinins act as vasodilators that interact with and enhance the action of prostaglandins while modulating the reninangiotensin system.[45] For example, kinins stimulate phospholipase A2 , and kininase (which controls the intrarenal kinin concentration) is blocked by ACE inhibitors. Two important intrarenal kinins, bradykinin and kallidin, appear to decrease the renal vasoconstriction induced by adrenergic hormones and angiotensin II.
The potential role of an endogenous natriuretic hormone was postulated for many years before ANP was identified. In 1972, Gorfinkel and coworkers[46] demonstrated a profound difference in the canine renal response to shock in that it depended on concomitant atrial pressure. Hypovolemic shock resulted in a rapid diminution in RBF
Indeed, an entire series of peptides with a similar precursor have been identified, with a core of 25 to 32 amino acids required for ANP-like activity. ANP is now referred to as human A-type natriuretic peptide in recognition of the existence of B-type natriuretic peptide (BNP) released from brain and cardiac ventricles and C-type natriuretic peptide released from the endothelium of major vessels.[49] Urodilatin is a natriuretic peptide produced in the lower urinary tract. Analogs have been developed and produced in recombinant form for exogenous administration, such as anaritide (derived from ANP) and nesiritide (derived from BNP). All these compounds induce arterial and venous dilation, increase RBF and GFR, and suppress the action of norepinephrine, angiotensin, and endothelin.
ANP is released from electron-dense granules in atrial myocytes in response to local wall stretch and increased atrial volume.[33] It dilates vascular smooth muscle through activation of guanylate cyclase and the formation of cyclic guanosine monophosphate (cGMP). At the phospholipase C-linked receptor, ANP competitively blocks norepinephrine and noncompetitively blocks angiotensin II, thus reversing vascular smooth muscle constriction. ANP causes a prompt, sustained increase in GFR and the glomerular filtration fraction, even when RBF is not increased or when arterial pressure is decreased. This effect suggests that it causes afferent arteriolar dilation with or without efferent arteriolar constriction. The increased GFR increases the filtered load of sodium, but natriuresis may be due to increased medullary blood flow, which washes out the concentration gradient.[50]
ANP appears to have a mutually antagonistic interaction with endothelin, the endogenous vasoconstrictor peptide produced by vascular endothelium.[33] It opposes the renin-angiotensin-aldosterone system on several fronts ( Fig. 20-19 ). ANP inhibits renin secretion and decreases angiotensin-stimulated aldosterone release. It also inhibits aldosterone release directly at the zona glomerulosa of the adrenal cortex and blocks the salt-retaining action of aldosterone at the distal tubule and collecting duct. Through cGMP activation, it inhibits NaCl reabsorption at the medullary portion of the collecting duct.[22] ANP also promotes diuresis by inhibiting AVP secretion from the posterior pituitary and antagonizing its effect on the antidiuretic V2 receptor in the collecting duct.
The renal protective role of endogenous ANP was further elucidated by Shannon and associates,[51] who noticed that patients undergoing mitral valve replacement had lower urine output after cardiac surgery than did those undergoing aortic valve replacement or coronary revascularization. They discovered that patients whose postoperative mean left atrial pressure declined by more than 7 mm Hg from
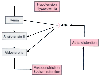 Figure 20-19
Interactions between atrial natriuretic peptide (ANP)
and the renin-angiotensin-aldosterone system. Hypotension or hypovolemia triggers
the release of renin from the afferent arteriole, thereby causing the formation of
angiotensin II, which stimulates the release of aldosterone from the adrenal cortex.
Angiotensin II and aldosterone cause vasoconstriction and sodium retention, ultimately
resulting in re-expansion of intravascular volume; this volume re-expansion causes
atrial distention, which triggers the release of ANP. ANP inhibits the release of
renin, renin's action on angiotensinogen to form angiotensin II, angiotensin-induced
vasoconstriction, stimulation of aldosterone secretion by angiotensin II, and the
action of aldosterone on the collecting duct. Thus, the actions of ANP promote vasodilation
and sodium excretion. Therapeutic administration of fluids to distend the atrium
and release ANP is an important intervention to curtail renal vasoconstriction and
sodium retention.
Figure 20-19
Interactions between atrial natriuretic peptide (ANP)
and the renin-angiotensin-aldosterone system. Hypotension or hypovolemia triggers
the release of renin from the afferent arteriole, thereby causing the formation of
angiotensin II, which stimulates the release of aldosterone from the adrenal cortex.
Angiotensin II and aldosterone cause vasoconstriction and sodium retention, ultimately
resulting in re-expansion of intravascular volume; this volume re-expansion causes
atrial distention, which triggers the release of ANP. ANP inhibits the release of
renin, renin's action on angiotensinogen to form angiotensin II, angiotensin-induced
vasoconstriction, stimulation of aldosterone secretion by angiotensin II, and the
action of aldosterone on the collecting duct. Thus, the actions of ANP promote vasodilation
and sodium excretion. Therapeutic administration of fluids to distend the atrium
and release ANP is an important intervention to curtail renal vasoconstriction and
sodium retention.
The dopaminergic (DA) receptor has two subtypes ( Table
20-2
).[52]
At the end organ, DA1
receptors occur not only on the renal and splanchnic vasculature but also on the
proximal tubule itself.[53]
Stimulation of the
DA1
| Receptor | DA1 | DA2 | β1 | β2 | α1 |
|---|---|---|---|---|---|
| Dopamine | +++ | ++ | ++ | ± | +++ |
| Dobutamine | 0 | 0 | +++ | ++ | ± |
| Dopexamine | ++ | + | ± | +++ | 0 |
| Fenoldopam | ++++ | 0 | 0 | 0 | 0 |
| DA, dopamine; 0, no activity; ±, minimal activity; ++-++++, increasing potency of adrenergic action. | |||||
 Figure 20-20
Correlation between left atrial pressure and plasma atrial
natriuretic peptide (ANP) in a group of patients undergoing cardiac surgery. A,
Significant correlation (r = 0.8, P
< .001) between absolute preoperative left atrial pressure and plasma ANP. B,
Significant correlation (r = 0.72, P
< .002) between the postoperative decrease in left atrial pressure and the postoperative
decrease in plasma ANP. ANF, atrial natriuretic factor (synonymous with ANP); Δ,
change. (From Shannon RP, Libby E, Elahi D, et al: Impact of acute reduction
in chronically elevated left atrial pressure on sodium and water excretion. Ann
Thorac Surg 46:430–437, 1988.)
Figure 20-20
Correlation between left atrial pressure and plasma atrial
natriuretic peptide (ANP) in a group of patients undergoing cardiac surgery. A,
Significant correlation (r = 0.8, P
< .001) between absolute preoperative left atrial pressure and plasma ANP. B,
Significant correlation (r = 0.72, P
< .002) between the postoperative decrease in left atrial pressure and the postoperative
decrease in plasma ANP. ANF, atrial natriuretic factor (synonymous with ANP); Δ,
change. (From Shannon RP, Libby E, Elahi D, et al: Impact of acute reduction
in chronically elevated left atrial pressure on sodium and water excretion. Ann
Thorac Surg 46:430–437, 1988.)
Neuronal DA2 receptors are found on the presynaptic terminal of postganglionic sympathetic nerves. Stimulation inhibits the release of norepinephrine from presynaptic vesicles, a mechanism analogous to stimulation of the presynaptic α2 -adrenergic receptor. Through inhibition of norepinephrine, DA2 receptor activation facilitates vasodilation.
Dopaminergic receptors form an integral component of the endogenous vasodilator-natriuresis system and are involved in maintenance of normal blood pressure. Endogenous dopamine appears to constitutively activate the DA2 receptor, which synergistically enhances activation of the DA1 receptor.[55] It acts as an autocrine and paracrine natriuretic factor by inhibiting tubular Na+ -K+ -ATPase activity, especially when sodium intake is increased.[56] It also opposes the antinatriuretic effects of norepinephrine and angiotensin II. Some evidence indicates that endogenous ANP also acts through the renal dopamine system by recruiting "silent" DA1 receptors from the interior of the cell toward the plasma membrane.[56] It has been suggested that decreased dopaminergic activity contributes to the pathogenesis of idiopathic edema, which is manifested as retention of salt and water in the upright position. [57]
Endogenous formation of nitric oxide is controlled by the enzyme nitric oxide synthase (NOS). NOS catalyzes hydroxylation of the nonessential amino acid L-arginine to L-citrulline. [58] Most actions of nitric oxide are mediated through its activation of soluble guanylate cyclase, which catalyzes the conversion of guanidine triphosphate to cGMP. cGMP has two major actions: relaxation of vascular smooth muscle and suppression of the inflammatory response. It inhibits leukocyte adhesion, platelet activation and aggregation, and cellular proliferation. cGMP is converted to GMP by phosphodiesterase I and V. Thus, the local action of nitric oxide can be enhanced by the administration of a selective phosphodiesterase V inhibitor such as sildenafil. Nitric oxide itself is rapidly inactivated by binding to intracellular heme and heme proteins (oxyhemoglobin, oxymyoglobin, guanylate cyclase, COX, cytochrome P450).
NOS has several distinct subtypes that determine the site and function of nitric oxide synthesis. Constitutive NOS is calcium and calmodulin dependent and releases small amounts of nitric oxide for short periods ("tonic" release). Constitutive NOS has two subtypes: neuronal NOS, which acts as a peripheral neurotransmitter and induces cerebral vasodilation, and endothelial NOS, which is found in the vascular endothelium and mediates the activity previously ascribed to endothelium-derived relaxing factor. The latter is an important modulator of systemic and pulmonary vascular resistance. In the kidney, endogenous nitric oxide preserves blood flow to the oligemic juxtamedullary cortex[59] and may also provide endogenous protection against ischemic and nephrotoxic renal insults. [60]
Inducible NOS is calcium and calmodulin independent and is induced by cytokines predominantly in
Goligorsky and Noiri[62] have framed the hypothesis that an imbalance between the expression and activity of constitutive and inducible NOS plays an important role in the pathophysiology of acute renal failure. In experimental models of sepsis, nonselective inhibitors of both constitutive and inducible NOS improve blood pressure but worsen overall perfusion, including renal perfusion. Selective inhibitors of inducible NOS show promise in suppressing severe inflammation and vasodilation while maintaining tonic perfusion to vital organs, including the kidneys.[63]
Adenosine, the endogenous degradation product of ATP, is produced by every mammalian cell type and is normally thought of as a potent vasodilator. However, in the kidney, it plays an essential role in regulating intrarenal blood flow by inducing outer cortical vasoconstriction and preserving juxtamedullary perfusion. This variance in function is explained by the identification of at least four subtypes of adenosine receptor: A1 , A2a , A2b , and A3 ( Table 20-3 ).[64] Activation of the A1 adenosine receptor induces outer cortical vasoconstriction; it also decreases renin release and inhibits diuresis and natriuresis. In contrast, A2a adenosine receptors increase medullary RBF and enhance renin release, diuresis, and natriuresis.
| Receptor | Agonist Function | Ischemic Injury |
|---|---|---|
| A1 | Outer cortical vasoconstriction | Highly protective |
|
|
Decreased renin release |
|
|
|
Inhibition of diuresis and natriuresis |
|
| A2a | Juxtamedullary vasodilation | Highly protective |
|
|
Increased renin release |
|
|
|
Promotion of diuresis and natriuresis |
|
| A2b | Unknown |
|
| A3 | Unknown | Potentiates injury |
In a series of studies in an in vivo rat model of ischemic acute renal failure, Lee and Emala[65] characterized the role of adenosine and its receptor subtypes in ischemic preconditioning. Pre-ischemic administration of adenosine as well as a selective A1 adenosine receptor agonist protected the kidney against global renal ischemic reperfusion injury. In contrast, pretreatment with selective A3 receptor activation potentiated ischemic injury. A selective A2a adenosine receptor agonist had the greatest renal protective effects, even if its administration was delayed until the early reperfusion period after termination of renal ischemia.[66]
It is conceivable that A1 adenosine receptor stimulation decreases renal oxygen consumption through a decrease in cortical blood flow, GFR, and sympathetic tone and that A2 adenosine receptor stimulation increases renal oxygen delivery through increased medullary blood flow. In addition, adenosine has cytoprotective properties, is the key mediator of ischemic preconditioning in the heart and brain, and is known to increase cellular resistance to ischemia in these organ systems. Pharmacologic development of a safe, specific A2a adenosine receptor agonist might provide specific protection against renal ischemic injury.
|
|
|
|
|
|
|
|
|
|
|
|
|