|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The end products of aerobic metabolism (oxidative phosphorylation) are CO2 and water, both of which are easily diffusible and lost from the body. The essential feature of hypoxia is the cessation of oxidative phosphorylation when mitochondrial PO2 falls below a critical level. Anaerobic pathways, which produce energy (ATP) inefficiently, are then used. The main anaerobic metabolites are hydrogen and latate ions, which are not easily excreted. They accumulate in the circulation, where they may be quantified in terms of the base deficit and the lactate-pyruvate ratio.
Because the various organs have different blood flow and O2
consumption rates, the manifestations and clinical diagnosis of hypoxia are usually
related to symptoms arising from the most vulnerable organ. This organ is
|
|
Hemodynamic Variable |
|
||||
|---|---|---|---|---|---|---|
| O2 Saturation (%) | HR | BP | SV | CO | SVR | Predominant Response |
| >80 | ↑ | ↑ | ↑ | ↑ | No change | Reflex, excitatory |
| 60–80 | ↑Baroreceptor | ↓ | No change | No change | ↓ | Local, depressant > reflex, excitatory |
| <60 | ↓ | ↓ | ↓ | ↓ | ↓ | Local, depressant |
| BP, systemic blood pressure; CO, cardiac output; HR, heart rate; SV, stroke volume; SVR, systemic vascular resistance; ↑, increase; ↓ = decrease. | ||||||
The cardiovascular response to hypoxemia[198] [199] [200] is a product of both reflex (neural and humoral) and direct effects ( Table 17-6 ). The reflex effects occur first and are excitatory and vasoconstrictive. The neuroreflex effects result from aortic and carotid chemoreceptor, baroreceptor, and central cerebral stimulation, and the humoral reflex effects result from catecholamine and reninangiotensin release. The direct local vascular effects of hypoxia are inhibitory and vasodilatory and occur late. The net response to hypoxia in a subject depends on the severity of the hypoxia, which determines the magnitude and balance between the inhibitory and excitatory components; the balance may vary according to the type and depth of anesthesia and the degree of preexisting cardiovascular disease.
Mild arterial hypoxemia (arterial saturation less than normal
but still 80% or higher) causes general activation of the sympathetic nervous system
and release of catecholamines. Consequently, the heart rate, stroke volume, ![]() T,
and myocardial contractility (as measured by a shortened pre-ejection period [PEP],
left ventricular ejection time [LVET], and a decreased PEP/LVET ratio) are increased
( Fig. 17-41
).[201]
Changes in systemic vascular resistance are usually slight. However, in patients
under anesthesia with β-blockers, hypoxia (and hypercapnia when present) may
cause circulating catecholamines to have only an α-receptor effect, the heart
may be unstimulated (even depressed by a local hypoxia effect), and systemic vascular
resistance may be increased. Consequently,
T,
and myocardial contractility (as measured by a shortened pre-ejection period [PEP],
left ventricular ejection time [LVET], and a decreased PEP/LVET ratio) are increased
( Fig. 17-41
).[201]
Changes in systemic vascular resistance are usually slight. However, in patients
under anesthesia with β-blockers, hypoxia (and hypercapnia when present) may
cause circulating catecholamines to have only an α-receptor effect, the heart
may be unstimulated (even depressed by a local hypoxia effect), and systemic vascular
resistance may be increased. Consequently, ![]() T
may be decreased in these patients. With moderate hypoxemia (arterial O2
saturation of 60% to 80%), local vasodilation begins to predominate and systemic
vascular resistance and blood pressure decrease, but the heart rate may continue
to be increased because of a systemic hypotension-induced stimulation of baroreceptors.
Finally, with severe hypoxemia (arterial saturation less than 60%), local depressant
effects dominate and blood pressure falls rapidly, the pulse slows, shock develops,
and the heart either fibrillates or becomes asystolic. Significant preexisting hypotension
will convert a mild hypoxemic hemodynamic profile into a moderate hypoxemic hemodynamic
profile and convert a moderate hypoxemic hemodynamic profile into a severe hypoxemic
T
may be decreased in these patients. With moderate hypoxemia (arterial O2
saturation of 60% to 80%), local vasodilation begins to predominate and systemic
vascular resistance and blood pressure decrease, but the heart rate may continue
to be increased because of a systemic hypotension-induced stimulation of baroreceptors.
Finally, with severe hypoxemia (arterial saturation less than 60%), local depressant
effects dominate and blood pressure falls rapidly, the pulse slows, shock develops,
and the heart either fibrillates or becomes asystolic. Significant preexisting hypotension
will convert a mild hypoxemic hemodynamic profile into a moderate hypoxemic hemodynamic
profile and convert a moderate hypoxemic hemodynamic profile into a severe hypoxemic
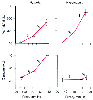 Figure 17-41
Changes in the minute ventilation and the circulation
of healthy awake humans during progressive isocapnic hypoxia and hyperoxic hypercapnia.
PETCO2
, end-tidal PCO2
;
PETO2
, end-tidal PO2
;
Figure 17-41
Changes in the minute ventilation and the circulation
of healthy awake humans during progressive isocapnic hypoxia and hyperoxic hypercapnia.
PETCO2
, end-tidal PCO2
;
PETO2
, end-tidal PO2
;
![]() , cardiac output; S1
, slope during the first phase of slowly increasing
ventilation and/or circulation; S2
, slope during the second phase of sharply
increasing ventilation and/or circulation. V̇E,
expired minute ventilation. (Redrawn from Serebrovskaya TV: Comparison
of respiratory and circulatory human responses to progressive hypoxia and hypercapnia.
Respiration 59:35, 1992.)
, cardiac output; S1
, slope during the first phase of slowly increasing
ventilation and/or circulation; S2
, slope during the second phase of sharply
increasing ventilation and/or circulation. V̇E,
expired minute ventilation. (Redrawn from Serebrovskaya TV: Comparison
of respiratory and circulatory human responses to progressive hypoxia and hypercapnia.
Respiration 59:35, 1992.)
Hypoxemia may also promote cardiac arrhythmias, which may in turn potentiate the already mentioned deleterious cardiovascular effects. Hypoxemia-induced arrhythmias may be caused by multiple mechanisms; the mechanisms are interrelated because they all cause a decrease in the myocardial O2 supply-demand ratio, which in turn increases myocardial irritability. First, arterial hypoxemia may directly decrease the myocardial O2 supply. Second, early tachycardia may result in increased myocardial O2 consumption, and decreased diastolic filling time may lead to decreased myocardial O2 supply. Third, early increased systemic blood pressure may cause an increased afterload on the left ventricle, which increases left ventricular O2 demand. Fourth, late systemic hypotension may decrease myocardial O2 supply because of decreased diastolic perfusion pressure. Fifth, coronary blood flow reserve may be exhausted by a late, maximally increased coronary blood flow as a result of maximal coronary vasodilation.[203] The level of hypoxemia that will cause cardiac arrhythmias cannot be predicted with certainty because the myocardial O2 supply-demand relationship in a given patient is not known (i.e., the degree of coronary artery atherosclerosis may not be known). However, if a myocardial area (or areas) become hypoxic or ischemic, or both, unifocal or multifocal premature ventricular contractions, ventricular tachycardia, and ventricular fibrillation may occur.
The cardiovascular response to hypoxia includes a number of other important effects. Cerebral blood flow increases (even if hypocapnic hyperventilation is present). Ventilation will be stimulated no matter why hypoxia exists (see Fig. 17-41 ). The pulmonary distribution of blood flow is more homogeneous because of increased pulmonary artery pressure. Chronic hypoxia causes an increased Hb concentration and a right-shifted oxy-Hb curve (as a result of either an increase in 2,3-DPG or acidosis), which tends to raise tissue PO2 .
The dangers associated with the inhalation of excessive O2 are multiple (see Chapter 70 ). Exposure to high O2 tension clearly causes pulmonary damage in healthy individuals.[204] [205] A dose-time toxicity curve for humans is available from a number of studies.[204] [205] [206] Because the lungs of normal human volunteers cannot be directly examined to determine the rate of onset and the course of toxicity, indirect measures such as the onset of symptoms have been used to construct dose-time toxicity curves. Examination of the curve indicates that 100% O2 should not be administered for more than 12 hours, 80% O2 should not be administered for more than 24 hours, and 60% O2 should not be administered for more than 36 hours.[204] [205] [206] No measurable changes in pulmonary function or blood-gas exchange occur in humans during exposure to less than 50% O2 , even for long periods.[206] Nevertheless, it is important to note that in the clinical setting, these dose-time toxicity relationships are often generally obscured[207] because of the complex multivariable nature of the clinical setting.
The dominant symptom of O2 toxicity in human volunteers is substernal distress, which begins as mild irritation in the area of the carina and may be accompanied by occasional coughing.[208] As exposure continues, the pain becomes more intense, and the urge to cough and to deep-breathe also becomes more intense. These symptoms will progress to severe dyspnea, paroxysmal coughing, and decreased vital capacity when the FIO2 has been 1.0 for longer than 12 hours. At this point, recovery of mechanical lung function usually occurs within 12 to 24 hours, but more than 24 hours may be required in some individuals.[206] As toxicity progresses, other pulmonary function studies such as compliance and blood gases deteriorate. Pathologically, in animals, the lesion progresses from tracheobronchitis (exposure for 12 hours to a few days), to involvement of the alveolar septa with pulmonary interstitial edema (exposure for a few days to 1 week), to pulmonary fibrosis of the edema (exposure for more than 1 week).[209]
Ventilatory depression may occur in patients who, by reason of drugs or disease, have been ventilating in response to a hypoxic drive. By definition, ventilatory depression resulting from removal of a hypoxic drive by
Absorption atelectasis was presented earlier (see the section "High Inspired Oxygen Concentration and Absorption Atelectasis"). Retrolental fibroplasia, an abnormal proliferation of the immature retinal vasculature of a prematurely born infant, can occur after exposure to hyperoxia. Extremely premature infants are most susceptible to retrolental fibroplasia (i.e., those less than 1.0 kg in birth weight and 28 weeks' gestation). The risk of retrolental fibroplasia exists whenever FIO2 causes PaO2 to be more than 80 mm Hg for more than 3 hours in an infant whose gestational age plus life age combined is less than 44 weeks. If the ductus arteriosus is patent, arterial blood samples should be drawn from the right radial artery (umbilical or lower extremity PaO2 is lower than the PaO2 to which the eyes are exposed because of ductal shunting of unoxygenated blood).
The mode of action of O2 toxicity in tissues is complex, but interference with metabolism seems to be widespread. Most importantly, many enzymes, particularly those with sulfhydryl groups, are inactivated by O2 -derived free radicals.[207] Neutrophil recruitment and release of mediators of inflammation occur next and greatly accelerate the extent of endothelial and epithelial damage and impairment of the surfactant systems.[207] The most acute toxic effect of O2 in humans is a convulsive effect, which occurs during exposure to pressures in excess of 2 atm absolute.
High inspired O2 concentrations can be of use therapeutically. Clearance of gas loculi in the body may be greatly accelerated by the inhalation of 100% O2 . Inhalation of 100% O2 creates a large nitrogen gradient from the gas space to the perfusing blood. As a result, nitrogen leaves the gas space and the space diminishes in size. Administration of O2 to remove gas may be used to ease intestinal gas pressure in patients with intestinal obstruction, decrease the size of an air embolus, and aid in the absorption of pneumoperitoneum, pneumocephalus, and pneumothorax.
The effects of CO2
on the cardiovascular system are
as complex as those of hypoxia. Like hypoxemia, hypercapnia appears to cause direct
depression of both cardiac muscle
| Anesthesia | Heart Rate | Contractility | Cardiac Output | Systemic Vascular Resistance |
|---|---|---|---|---|
| Conscious | ++ | ++ | +++ | - |
| Nitrous oxide | 0 | + | ++ | -- |
| Halothane | 0 | + | + | - |
| Enflurane | + | + | + | --- |
| Isoflurane | ++ | ++ | +++ | - |
| +, <10% increase; ++, 10% to 25% increase; +++, >25% increase; 0, no change; -, <10% decrease; -- 10% to 25% decrease; ---, >25% decrease; MAC, minimum alveolar concentration for adequate anesthesia in 50% of subjects. | ||||
Table 17-7
summarizes the interaction of anesthesia with hypercapnia in humans; increased ![]() T
and decreased systemic vascular resistance should be emphasized.[210]
[211]
The increase in
T
and decreased systemic vascular resistance should be emphasized.[210]
[211]
The increase in ![]() T
is most marked during anesthesia with drugs that enhance sympathetic activity and
least marked with halothane and nitrous oxide. The decrease in systemic vascular
resistance is most marked during enflurane anesthesia and hypercapnia. Hypercapnia
is a potent pulmonary vasoconstrictor even after the inhalation of 3% isoflurane
for 5 minutes.[210]
T
is most marked during anesthesia with drugs that enhance sympathetic activity and
least marked with halothane and nitrous oxide. The decrease in systemic vascular
resistance is most marked during enflurane anesthesia and hypercapnia. Hypercapnia
is a potent pulmonary vasoconstrictor even after the inhalation of 3% isoflurane
for 5 minutes.[210]
Arrhythmias have been reported in unanesthetized humans during acute hypercapnia, but they have seldom been of serious import. A high PaCO2 level is, however, more dangerous during general anesthesia. With halothane anesthesia, arrhythmias frequently occur above a PaCO2 arrhythmic threshold that is often constant for a particular patient. Furthermore, halothane, enflurane, and isoflurane have been shown to prolong the QTC interval in humans, thereby increasing the risk for torsades de pointes ventricular tachycardia, which in turn is notorious for decompensating into ventricular fibrillation.[212]
The maximum stimulatory respiratory effect is attained by a PaCO2 of about 100 mm Hg. With a higher PaCO2 , stimulation is reduced, and at extremely high levels, respiration is depressed and later ceases altogether. The PCO2 ventilation-response curve is generally displaced to the right, and its slope is reduced by anesthetics and other depressant drugs.[213] With profound anesthesia, the response curve may be flat or even sloping downward, and CO2 then acts as a respiratory depressant. In patients with ventilatory failure, CO2 narcosis occurs when PaCO2 rises to more than 90 to 120 mm Hg. A 30% CO2
Quite apart from the effect of CO2 on ventilation, it exerts two other important effects that influence the oxygenation of the blood. [116] First, if the concentration of nitrogen (or other inert gas) remains constant, the concentration of CO2 in alveolar gas can increase only at the expense of O2 , which must be displaced. Thus, PAO2 and PaO2 may decrease. Second, hypercapnia shifts the oxy-Hb curve to the right, thereby facilitating tissue oxygenation.[98]
Chronic hypercapnia results in increased resorption of bicarbonate by the kidneys, which further raises the plasma bicarbonate level and constitutes a secondary or compensatory metabolic alkalosis. The decrease in renal resorption of bicarbonate in patients with chronic hypocapnia results in a further fall in plasma bicarbonate and produces a secondary or compensatory metabolic acidosis. In each case, arterial pH returns toward the normal value, but the bicarbonate ion concentration departs even further from normal.
Hypercapnia is accompanied by leakage of potassium from cells into plasma. Much of the potassium comes from the liver, probably from glucose release and mobilization, which occur in response to the rise in plasma catecholamine levels. [216] Because the plasma potassium level takes an appreciable time to return to normal, repeated bouts of hypercapnia at short intervals result in a stepwise rise in plasma potassium. Finally, hypercapnia can predispose the patient to other complications in the operating room (e.g., the oculocephalic response is far more common during hypercapnia than during eucapnia).[217]
In this section, hypocapnia is considered to be produced by passive
hyperventilation (by the anesthesiologist or ventilator). Hypocapnia may cause a
decrease in ![]() T by three separate mechanisms.
First, if it is present, an increase in intrathoracic pressure will decrease cardiac
output. Second, hypocapnia is associated with withdrawal of sympathetic nervous
system activity, and such withdrawal can decrease the inotropic state of the heart.
Third, hypocapnia can increase pH, and the increased pH can decrease ionized calcium,
which may in turn decrease the inotropic state of the heart. Hypocapnia with alkalosis
also shifts the oxy-Hb curve to the left, which increases Hb affinity for O2
and thus impairs O2
unloading at the tissue level. The decrease in peripheral
flow and the impaired ability to unload O2
to the tissues are compounded
by an increase in whole-body O2
consumption as a result of increased pH-mediated
uncoupling of oxidation from phosphorylation.[218]
A PaCO2
of 20 mm Hg will increase tissue
O2
consumption by 30%. Consequently, hypocapnia may simultaneously increase
tissue O2
demand and decrease tissue O2
supply. Thus, to have
the same amount of O2
delivery to the tissues,
T by three separate mechanisms.
First, if it is present, an increase in intrathoracic pressure will decrease cardiac
output. Second, hypocapnia is associated with withdrawal of sympathetic nervous
system activity, and such withdrawal can decrease the inotropic state of the heart.
Third, hypocapnia can increase pH, and the increased pH can decrease ionized calcium,
which may in turn decrease the inotropic state of the heart. Hypocapnia with alkalosis
also shifts the oxy-Hb curve to the left, which increases Hb affinity for O2
and thus impairs O2
unloading at the tissue level. The decrease in peripheral
flow and the impaired ability to unload O2
to the tissues are compounded
by an increase in whole-body O2
consumption as a result of increased pH-mediated
uncoupling of oxidation from phosphorylation.[218]
A PaCO2
of 20 mm Hg will increase tissue
O2
consumption by 30%. Consequently, hypocapnia may simultaneously increase
tissue O2
demand and decrease tissue O2
supply. Thus, to have
the same amount of O2
delivery to the tissues, ![]() T
or tissue perfusion has to increase at a time when it may not be possible to do so.
The cerebral effects of hypocapnia may be related to a state of cerebral acidosis
and hypoxia because hypocapnia may cause a selective reduction in cerebral blood
flow and also shifts the oxy-Hb curve to the left.[219]
T
or tissue perfusion has to increase at a time when it may not be possible to do so.
The cerebral effects of hypocapnia may be related to a state of cerebral acidosis
and hypoxia because hypocapnia may cause a selective reduction in cerebral blood
flow and also shifts the oxy-Hb curve to the left.[219]
Hypocapnia may cause V̇A/![]() abnormalities by inhibiting HPV or by causing bronchoconstriction and decreased CL.
Finally, passive hypocapnia promotes apnea.
abnormalities by inhibiting HPV or by causing bronchoconstriction and decreased CL.
Finally, passive hypocapnia promotes apnea.
|
|
|
|
|
|
|
|
|
|
|
|
|