|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As defined by Wright,[226]
pharmacokinetics
and pharmacodynamics are "... empirical mathematical model[s] ... that [describe]
drug effect time course after administration." In pharmacokinetic modeling, the
concept of "compartments" represents different organs/tissues grouped together on
the basis of their blood perfusion (high or low). After injection into the circulation,
the concentration of a neuromuscular blocker in plasma decreases rapidly at first,
then more slowly ( Fig. 13-15
).
The shape of this curve is determined by the processes of distribution and elimination.
Classically, this curve is divided into an initial (distribution) phase and a terminal
(elimination) phase. This curve can be represented mathematically by biexponential
or triexponential equations[223]
in the form
Concentration (at time t) = Ae−αt
+ Be−βt
(+ Pe−πt
)
These multiexponential equations express the concept of drug being distributed between
two or three theoretical compartments.
Figure 13-16 illustrates the classic model whereby drug is administered intravenously into a central compartment with volume V1 and is distributed and eliminated only from this compartment. Drug is distributed very rapidly throughout this central compartment, which includes the plasma volume and the organs of elimination (e.g., in the case of neuromuscular blockers, the kidneys and liver). The "k" terms are the rate constants for drug movement between compartments in the direction of the arrows. The peripheral compartments (usually one or two
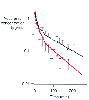 Figure 13-15
Disappearance of vecuronium from plasma after a single
bolus dose of 0.2 mg/kg, as illustrated by a semilogarithmic plot of the mean concentration
versus time for patients with normal hepatic function (filled
circles) and cirrhotic patients (open circles).
Error bars are the SD for that value. (Redrawn from Lebrault C, Berger
JL, D'Hollander AA, et al: Pharmacokinetics and pharmacodynamics of vecuronium [ORG
NC 45] in patients with cirrhosis. Anesthesiology 62:601–605, 1985.)
Figure 13-15
Disappearance of vecuronium from plasma after a single
bolus dose of 0.2 mg/kg, as illustrated by a semilogarithmic plot of the mean concentration
versus time for patients with normal hepatic function (filled
circles) and cirrhotic patients (open circles).
Error bars are the SD for that value. (Redrawn from Lebrault C, Berger
JL, D'Hollander AA, et al: Pharmacokinetics and pharmacodynamics of vecuronium [ORG
NC 45] in patients with cirrhosis. Anesthesiology 62:601–605, 1985.)
Initially, the drug concentration in the central compartment (plasma concentration) will exceed that in the peripheral compartment (tissue concentration), and drug
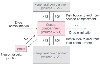 Figure 13-16
Schematic representation of drug disposition into different
compartments. These compartments are mathematical concepts only and do not represent
real physiologic spaces. The effect compartment in this case would be the neuromuscular
junction for computational purposes; it has infinitesimal volume. The terms knm
are the rate constants for drug movement, in the direction of the arrow,
between these theoretical compartments. See text for further discussion.
Figure 13-16
Schematic representation of drug disposition into different
compartments. These compartments are mathematical concepts only and do not represent
real physiologic spaces. The effect compartment in this case would be the neuromuscular
junction for computational purposes; it has infinitesimal volume. The terms knm
are the rate constants for drug movement, in the direction of the arrow,
between these theoretical compartments. See text for further discussion.
Volume of distribution is the volume to which the drug has distributed when the processes of distribution and elimination are in equilibrium. Elimination is represented by the variable plasma clearance, that is, the volume of plasma from which drug is irreversibly and completely eliminated per unit time. For most nondepolarizing neuromuscular blockers, the process of distribution is more rapid than that of elimination, and the initial rapid decline in plasma concentration is due primarily to distribution of the drug to tissues. An exception to this rule is mivacurium, which has such rapid clearance, because of metabolism by butyrylcholinesterase, that elimination is the principal determinant of the initial decline in plasma concentration.[228]
After the initial process of drug distribution to tissues, the plasma concentration falls more slowly (terminal phase). The rate of decrease in plasma concentration during this terminal phase is often expressed in terms of elimination half-life, which equals the natural logarithm of 2 divided by the rate constant of decline (i.e., the slope of the terminal phase). During this terminal phase, the tissue drug concentration exceeds that of plasma, and the rate of decrease in plasma concentration is determined by two factors: the rate at which drug can move from tissues back to plasma and clearance of drug from plasma. In classic theory for neuromuscular blockers, drug can move rapidly from tissues to plasma, and elimination from plasma (clearance) is the rate-limiting step. For this reason, the terminal portion of the curve is often termed the elimination phase, even though distribution of drug from tissues to plasma is occurring continually throughout. Volume of distribution can also influence the terminal portion of the curve; the greater the volume of distribution, the slower the decline in plasma concentration.
The neuromuscular blockers are polar drugs, and their volume of distribution is classically thought to be limited to a volume roughly equivalent to a portion of the extracellular fluid space, specifically, 150 to 450 mL/kg (see Table 13-14 and Table 13-15 ).[223] With this model of drug distribution, the potential rate of drug movement from tissues to plasma exceeds the rate of elimination, and plasma clearance is the process that limits the rate of decline in plasma drug concentration. However, some evidence has shown that neuromuscular blockers are distributed more widely, into tissues with low blood flow (e.g., connective tissue),[229] and the true volume of distribution of dTc has been estimated to be as high as 3.4 L/kg and the elimination half-life as long as 40 hours (compare with values in Table 13-14 and Table 13-15 ). [230] Because the rate of drug movement from such tissues is less than that of plasma clearance, this becomes the rate-limiting step in the rate of decline in plasma drug concentration. This phase only becomes obvious when drug is administered for many days or when sampling is continued for 24 to 96 hours after drug administration. In normal operating room use of neuromuscular blockers, the amount of drug being distributed to this compartment does not affect clinical response to the drug. In conditions in which clearance of the neuromuscular blocker is reduced, such as renal or hepatic disease, it is the terminal portion of the plasma concentration-versus-time curve that is most affected (see Fig. 13-15 ).[231] The rate of decline in plasma concentration is slowed, and recovery from paralysis is potentially delayed.[231] In conditions associated with an increased distribution volume, such as renal or hepatic disease, early plasma concentrations of drug may be less than those observed when organ function is normal ( Fig. 13-17 ). With a greater volume of distribution, the plasma concentration should be less whereas the total amount of a drug would be greater (see Fig. 13-17 ). Decreased protein binding of a drug results in a larger distribution volume, but because the degree of protein binding of neuromuscular blockers is low, changes in protein binding will have minimal effect on their distribution.[232]
Recovery of neuromuscular function takes place as plasma concentrations decline. The greater part of this decline occurs primarily because of distribution. Thus, processes that primarily affect elimination of the drug, such as renal failure, may not be associated with a prolonged duration of block.[233] [234] However, as recovery comes to rely more on drug elimination than distribution, that is, recovery from 25% to 75% or more or after the administration of larger or repeated doses, the duration of action may be prolonged.[227] [235]
After injection of a neuromuscular blocker, the plasma drug concentration immediately starts to decrease. The effect (neuromuscular blockade) takes approximately 1 minute to begin, increases initially, and does not begin to recover for many more minutes despite continually decreasing plasma concentrations of drug. This discrepancy between plasma concentration and drug effect occurs
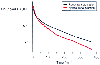 Figure 13-17
"Average" plasma concentration (Cp) versus time after
0.6 mg/kg rocuronium in patients with normal renal function or patients undergoing
renal transplantation. (Redrawn from Szenohradszky J, Fisher DM, Segredo
V, et al: Pharmacokinetics of rocuronium bromide [ORG 9426] in patients with normal
renal function or patients undergoing cadaver renal transplantation. Anesthesiology
77:899–904, 1992.)
Figure 13-17
"Average" plasma concentration (Cp) versus time after
0.6 mg/kg rocuronium in patients with normal renal function or patients undergoing
renal transplantation. (Redrawn from Szenohradszky J, Fisher DM, Segredo
V, et al: Pharmacokinetics of rocuronium bromide [ORG 9426] in patients with normal
renal function or patients undergoing cadaver renal transplantation. Anesthesiology
77:899–904, 1992.)
|
|
|
Adductor Pollicis |
|
|
|---|---|---|---|---|
|
|
Study Group | CE50 * (ng/mL) | ke0 † (min-1 ) | Reference |
| Mivacurium |
|
|
|
|
| Central link |
|
57 | 0.169 | [236] |
| Peripheral link |
|
130 | 0.101 | [236] |
| Rocuronium | Adult female | 684 | 0.329 | [237] |
|
|
Propofol-remifentanil anesthesia |
|
|
|
|
|
Standard model |
|
|
|
|
|
Recirculatory model | 876 | 0.129 | [237] |
|
|
Volunteers | 3510 | 0.405 | [238] |
|
|
Propofol-fentanyl anesthesia |
|
|
|
|
|
Infants | 1190 | 0.25 | [239] |
|
|
Children | 1650 | 0.32 | [239] |
| Cisatracurium | Adults | 126–158 | 0.07–0.09 | [240] |
| 0.075–3.0 mg/kg | Propofol-fentanyl anesthesia |
|
|
|
| Atracurium | Infants | 363 | 0.19 | [241] |
|
|
Children | 444 | 0.16 | [241] |
|
|
Young adults | 449 | 0.13 | [242] |
|
|
Elderly adults | 436 | 0.12 | [242] |
|
|
Standard model | 359 | 0.12 | [243] |
|
|
Threshold model | 357 | 0.12 | [243] |
|
|
Young adults | 669 ‡ | 0.07 | [244] |
|
|
Burn patients | 2270 ‡ | 0.10 | [244] |
|
|
No succinylcholine | 454 ‡ | 0.07 | [153] |
|
|
After succinylcholine | 305 ‡ | 0.09 | [153] |
| Vecuronium | Young adults | 94 | — | [245] |
|
|
Young adults | 92 | 0.17 | [246] |
|
|
Elderly adults | 106 | 0.17 | [246] |
| d-Tubocurarine | Normal renal function | 370 | 0.13 | [224] |
|
|
Renal failure | 380 | 0.16 | [224] |
|
|
Halothane 0.5%–0.7% | 360 ‡ | 0.09 ‡ | [247] § |
|
|
Halothane 1.0%–1.2% | 220 ‡ | 0.12 | [247] § |
|
|
Narcotic anesthesia | 600 ‡ | 0.15 ‡ | [247] § |
| Pancuronium | Young adults | 88 | — | [245] |
To overcome this problem, pharmacodynamic models have been developed to incorporate a factor for the delay caused by drug diffusion to and from the neuromuscular junction.[153] [224] [236] [237] [238] [239] [240] [241] [242] [243] [244] [245] [246] [247] This factor, ke0 , is the rate constant for drug equilibration between plasma and the neuromuscular junction. By measuring plasma drug concentrations and neuromuscular blockade duringboth the onset and recovery phases and by using the technique of simultaneous pharmacokinetic/pharmacodynamic modeling, it is possible to collapse the hysteresis in the plasma concentration-effect curve, estimate actual neuromuscular junction drug concentrations, and derive true concentration-effect relationships (CE50 and ke0 ) for the neuromuscular blockers ( Table 13-6 ).[224]
|
|
|
|
|
|
|
|
|
|
|
|
|