|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The major cause of an abnormal physiologic stress response in surgical intensive care is sepsis (see Chapter 56 and Chapter 74 ). Sepsis is the host's response to infection, which imposes another stress on the host in addition to the stress imposed by trauma alone. As expected, this leads to a continuum of responses with increasing severity. Ultimately, the patient improves and returns to the normal stress response or continues to deteriorate. The complications of sepsis lead to a condition described clinically as a sequential failure of the major organ systems of the body.[13] [26] [27] [28] In operations for intra-abdominal sepsis, multiple-system organ failure occurs in 30% to 50% of cases and is the major cause of death of patients with sepsis.
Between 3 and 4 days after the septic episode, patients enter a phase characterized by persistent hypermetabolism, which can last weeks. These patients are hemodynamically stable, demonstrating an increased cardiac output at 10 to 12 L/min. In contrast to the stress response, the total peripheral resistance falls, indicative of a hyperdynamic cardiovascular state. The oxygen consumption increases approximately twofold above normal ( Fig. 77-11 ). The respiratory gas quotient falls to 0.8 to 0.85, indicative of a greater reliance on fats for oxidative energy production. In general, there is marked increase in metabolic demand and work relative to the resting, fasting human. The plasma lactate level is increased into the 2 to 3 mg/dL range. However, this increase in lactate is not accompanied by a metabolic acidosis. The plasma pyruvate concentrations also rise such that the lactate-to-pyruvate ratio in the arterial and femoral vein plasma is within normal limits, implying a normal cytosolic redox potential.[29] Without any overt injury to specific organ systems, the level of plasma enzymes used as indicators of ischemic injury (i.e., serum glutamic oxaloacetic transaminase, creatine phosphokinase, lactate dehydrogenase, and ornithine transcarbamylase) are not increased. Biopsies of skeletal muscle reveal adequate levels of high-energy phosphates, also indicative of adequate blood flow. Under this condition, energy production and use are consistent with adequate oxygen supply, and metabolically insignificant amounts of hypoxia-driven metabolic alterations are observed. Instead, other mechanisms or abnormalities besides inadequate oxygen delivery are involved in the regulation of plasma metabolites and fluxes in sepsis.
As the septic processes progress, hemodynamic and metabolic dysfunctions are observed.[12] In particular, the
 Figure 77-11
Cardiovascular and metabolic responses after trauma and
sepsis in 10 septic and 16 nonseptic patients. Mean values over the postinjury course
for cardiac index (CI) and oxygen consumption (ψO2
)
are shown for trauma patients who developed sepsis (ST) and compared with those who
had nonseptic courses. Notice the increase in CI and ψO2
in ST patients as sepsis becomes established between 5 and 7 days after injury.
(Adapted from Sganga G, Siegel JH, Brown C, et al: Reprioritization of hepatic
plasma protein release in trauma and sepsis. Arch Surg 120:187, 1985.)
Figure 77-11
Cardiovascular and metabolic responses after trauma and
sepsis in 10 septic and 16 nonseptic patients. Mean values over the postinjury course
for cardiac index (CI) and oxygen consumption (ψO2
)
are shown for trauma patients who developed sepsis (ST) and compared with those who
had nonseptic courses. Notice the increase in CI and ψO2
in ST patients as sepsis becomes established between 5 and 7 days after injury.
(Adapted from Sganga G, Siegel JH, Brown C, et al: Reprioritization of hepatic
plasma protein release in trauma and sepsis. Arch Surg 120:187, 1985.)
It is in the setting of the stable hyperdynamic cardiovascular condition that patients need the maximal nutritional support to prevent sustained starvation from limiting recovery.[30] The septic insult results in pathologic alterations in glucose, fatty acid, and amino acid (protein) metabolism ( Fig. 77-12 ). Virtually no organ system is spared from the metabolic alterations in sepsis. The pattern of alterations in the plasma level of these fuels is superimposed on changes in the plasma level of hormones. Changes in carbohydrate metabolism after severe trauma and during sepsis include hyperglycemia, increased gluconeogenesis with an increased output of glucose from the liver, hyperlactatemia, and insulin resistance. The metabolic alterations that occur in sepsis are numerous, and all probably contribute to a cascade of secondary alterations that result from the initial traumatic or septic episode. Collectively, these changes lead to the observed plasma alteration in hormones and substrates in the critically ill patient. The metabolic changes observed are likely to result from changes in hormone concentrations or expression of inflammatory or immunologic mediators.
Sepsis increases glucose turnover by stimulating hepatic glucose production and by augmenting peripheral glucose use.[31] [32] Gluconeogenesis represents the major pathway for lactate clearance, and enhanced rates of gluconeogenesis are associated with a twofold to threefold increase in net lactate extraction by the liver during sepsis. [33] [34] In injured patients, endogenous glucose production is elevated twofold to threefold above the normal basal level. Two or three times as much infused glucose may be required to suppress glucose production in the injured patient. In contrast, sepsis appears to minimize the effectiveness of exogenous glucose in suppressing glucose production. The response to glucose in any patient can be determined by recording the
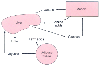 Figure 77-12
Interorgan substrate fluxes in response to sepsis.
Figure 77-12
Interorgan substrate fluxes in response to sepsis.
Sepsis also increases glucose use, as measured by lactate and alanine production. However, the increased glucose uptake is not accompanied by a corresponding increase in glucose oxidation. Instead, the glucose carbon is released
 Figure 77-13
Glucose concentration and glucose clearance (i.e., glucose
uptake/glucose concentration) in postoperative patients on total parenteral nutrition.
There is a progressive rise in glucose clearance, and after 6 days of infusion,
the glucose concentration is significantly lower than at 2 hours of infusion, even
though the infusion rate is much higher at 6 days. Glucose production is suppressed
in normal volunteers by exogenous glucose infusion. During the first 2 hours of
glucose infusion, suppression of glucose production appears to be of predominant
importance in maintaining normal blood glucose concentration. Maximal suppression
can be obtained at glucose infusion rates well below rates conventionally administered
in total parenteral nutrition. (Adapted from Wolfe RR: Regulation of glucose
metabolism. In Burke JF [ed]: Surgical Physiology.
Philadelphia, WB Saunders, 1983, p 75.)
Figure 77-13
Glucose concentration and glucose clearance (i.e., glucose
uptake/glucose concentration) in postoperative patients on total parenteral nutrition.
There is a progressive rise in glucose clearance, and after 6 days of infusion,
the glucose concentration is significantly lower than at 2 hours of infusion, even
though the infusion rate is much higher at 6 days. Glucose production is suppressed
in normal volunteers by exogenous glucose infusion. During the first 2 hours of
glucose infusion, suppression of glucose production appears to be of predominant
importance in maintaining normal blood glucose concentration. Maximal suppression
can be obtained at glucose infusion rates well below rates conventionally administered
in total parenteral nutrition. (Adapted from Wolfe RR: Regulation of glucose
metabolism. In Burke JF [ed]: Surgical Physiology.
Philadelphia, WB Saunders, 1983, p 75.)
 Figure 77-14
Gluconeogenic response to trauma in 10 septic and 16
nonseptic patients. The mean values for plasma glucose and alanine in patients with
sepsis after trauma (ST) are compared with those of patients with nonseptic post-trauma
courses (NST). Notice the early rise in the plasma glucose level associated with
an increase in plasma alanine in ST patients with progressive increase as the septic
course evolves. (Adapted from Vary TC, Siegel JH: Sepsis, abnormal metabolic
control and multiple organ failure syndrome. In
Siegel JH [ed]: Trauma: Emergency Surgery and Critical Care. New York, Churchill
Livingstone, 1987, p 411.)
Figure 77-14
Gluconeogenic response to trauma in 10 septic and 16
nonseptic patients. The mean values for plasma glucose and alanine in patients with
sepsis after trauma (ST) are compared with those of patients with nonseptic post-trauma
courses (NST). Notice the early rise in the plasma glucose level associated with
an increase in plasma alanine in ST patients with progressive increase as the septic
course evolves. (Adapted from Vary TC, Siegel JH: Sepsis, abnormal metabolic
control and multiple organ failure syndrome. In
Siegel JH [ed]: Trauma: Emergency Surgery and Critical Care. New York, Churchill
Livingstone, 1987, p 411.)
Impairment in glucose oxidation in conjunction with normal or increased lactate, alanine, or pyruvate production suggests a specific inhibition of the PDH reaction. This impairment can be caused by the decreased formation of pyruvate or the entry of glucose carbon into the tricarboxylic acid (TCA) cycle. Estimates of glucose recycling[31] [32] [33] [34] suggest that the rate of pyruvate formation from glucose is probably not rate limiting for glucose oxidation. Entry of glucose carbon into the TCA cycle may be limiting for glucose oxidation in sepsis. Some studies have identified a specific inhibition of the PDH complex in sepsis.
Endotoxin administration and bacterial infusions stimulate glucose uptake and phosphorylation in skeletal muscle.[35] [36] [37] The increased glucose uptake and phosphorylation is associated with a twofold increase in the glucose-6-phosphate content.[38] The mechanisms responsible for these changes are unknown. Glucose entry into muscle is facilitated by specific transport proteins. At least five isoforms of the facultative glucose transporter have been described, and two of these isoforms, GLUT-1 and GLUT-4, are present in skeletal muscle. The GLUT-1 isoform is present in a wide variety of tissues and mediates basal glucose uptake.[39] In contrast to GLUT-1, the GLUT-4 isoform is expressed only in those tissues in which glucose uptake is facilitated by insulin.[39] In skeletal muscle, glucose uptake is the rate-limiting step in glucose use. Sustained increases in glucose transport activity result from an increase in the number of cell surface glucose transporters.[39] Some studies have demonstrated an increased amount (approximately 70%) of GLUT-1 protein in skeletal muscle from septic animals compared with controls.[38] The increased GLUT-1 transporter expression correlates with enhanced basal (non-insulin-stimulated) glucose transport after endotoxin administration or sepsis.[40] [41] The significance of this observation is that non-insulin-mediated glucose uptake accounts for 75% to 85% of the total glucose disposal in the basal postabsorptive state.[39] The increased glucose transport in sepsis may result in part from an increased GLUT-1 transporter protein content.
There is a specific inhibition of the PDH complex in sepsis, and the degree of impairment depends on the severity of the septic episode.[42] [43] Because decreased PDH activity is associated with decreased pyruvate oxidation, the inhibition of the complex during sepsis may provide a biochemical explanation for the shift in skeletal muscle glucose metabolism in sepsis. The inhibition of the PDH complex results from an increased phosphorylation, catalyzed by the PDH kinase. The effect of sepsis to increase PDH kinase activity is mediated by an increase in the acetyl CoA/CoA concentration ratio.[42] In muscle tissue, the acetyl CoA/CoA concentration is a sensitive index of the availability of metabolic fuels to be oxidized by the TCA cycle. The ratio is increased when noncarbohydrate fuels are the major oxidative substrate. Because the acetyl CoA/CoA ratio is increased in skeletal muscle from septic rats,
Regulation of the PDH complex makes a significant contribution to glucose homeostasis during sepsis. In experimental septic animal models, activation of the PDH complex with dichloroacetate decreases skeletal muscle, liver, and plasma lactate, pyruvate, and alanine concentrations[48] [49] [50] [51] [52] ( Fig. 77-15 ). Concomitant with the decrease in plasma gluconeogenic precursors, the rates of glucose production and glucose turnover were reduced to nonseptic values. [49]
Typically, sepsis is characterized by a decreased level of the thyroid hormone triiodothyronine, whereas the levels of other stress hormones (i.e., cortisol, epinephrine, and glucagon) are elevated. Glucagon levels rise to an extraordinarily high level. Although the rise in glucagon is accompanied by a rise in immunoassayable insulin, the insulin-to-glucagon ratio is reversed compared with the postabsorptive state. This reversal of the insulin-to-glucagon ratio may be responsible in part for the accelerated rate of glucose production by the liver in sepsis. Insulin and glucagon have immediate and delayed effects on hepatic glucose metabolism. The immediate effects of glucagon on hepatic glucose production are seen
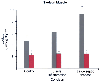 Figure 77-15
Effect of dichloroacetate in skeletal muscle. (Adapted
from Vary TC, Siegel JH: Sepsis, abnormal metabolic control and multiple organ failure
syndrome. In Siegel JH [ed]: Trauma: Emergency
Surgery and Critical Care. New York, Churchill Livingstone, 1987, p 411.)
Figure 77-15
Effect of dichloroacetate in skeletal muscle. (Adapted
from Vary TC, Siegel JH: Sepsis, abnormal metabolic control and multiple organ failure
syndrome. In Siegel JH [ed]: Trauma: Emergency
Surgery and Critical Care. New York, Churchill Livingstone, 1987, p 411.)
Catecholamines also stimulate glycogenolysis and gluconeogenesis. Plasma levels of catecholamines—epinephrine and norepinephrine—rise progressively with increasing severity of injury. Concentrations of epinephrine and norepinephrine rise to levels considered high enough to produce metabolic changes. In this regard, plasma epinephrine concentrations are more important for stimulating hyperglycemia than is the severity of the injury itself. The stimulation of hepatic output of glucose by epinephrine involves the breakdown of glycogen with the release of glucose. It appears that catecholamines activate glycogenolysis more effectively through a cAMP-α-independent mechanism involving α-receptors, rather than through cAMP production by stimulation of β-receptors. The α-adrenergic activation of glycogenolysis occurs because of an increase in phosphorylase a, resulting from stimulation of a Ca2+ -sensitive phosphorylase kinase. However, changes in the hormonal milieu are not solely responsible for enhanced gluconeogenesis in sepsis. Unlike other pathologic conditions, such as starvation and diabetes, in sepsis, gluconeogenesis is not suppressed by infusion of glucose.[31] [53] The failure of glucose to limit hepatic gluconeogenesis has been proposed to occur as a result of enhanced and continual delivery of gluconeogenic precursors from peripheral tissues.
|
|
|
|
|
|
|
|
|
|
|
|
|