|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In addition to its role in locomotion, skeletal muscle (by virtue of its mass in relationship to body weight) represents the major reservoir of amino acids. Some of these amino acids, including alanine, serine, and glycine, are important substrates for gluconeogenesis in liver and kidney. Protein wasting is a general feature of trauma, sepsis, and burns. The earliest recognizable disturbance in protein metabolism in the injury process is excessive urea excretion, resulting in a loss of nitrogen from the body.[12] [30] [31] [54] [55] Much of this nitrogen loss originates from protein in skeletal muscle. This catabolic phase is an intrinsic response to
The rate of muscle protein breakdown increases to a greater degree than that of the whole body, with the contribution of muscle proteolysis to whole-body protein degradation nearly doubling. The measurement of nitrogen excretion by the kidney can be used as an estimate of protein breakdown because the nitrogen excreted is derived from amino acids released by proteolysis. The increased proteolysis results in the release of amino acids from structural protein stores, particularly in skeletal muscle. The
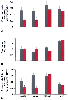 Figure 77-16
Effect of sepsis on the rate of protein synthesis (A),
RNA concentration (B), and translational efficiency (C) in gastrocnemius (gastroc),
psoas, soleus, and heart muscles. Open bars represent
control animals, shaded bars represent animals with
septic abscess (5 days after fecal-agar pellet infected with Escherichia
coli plus Bacteroides fragilis implanted
intraperitoneally.) (Adapted from Vary TC, Kimball SR: Sepsis-induced changes
in protein synthesis: Differential effects on fast and slow-twitch muscles. Am
J Physiol 262:C1513, 1992.)
Figure 77-16
Effect of sepsis on the rate of protein synthesis (A),
RNA concentration (B), and translational efficiency (C) in gastrocnemius (gastroc),
psoas, soleus, and heart muscles. Open bars represent
control animals, shaded bars represent animals with
septic abscess (5 days after fecal-agar pellet infected with Escherichia
coli plus Bacteroides fragilis implanted
intraperitoneally.) (Adapted from Vary TC, Kimball SR: Sepsis-induced changes
in protein synthesis: Differential effects on fast and slow-twitch muscles. Am
J Physiol 262:C1513, 1992.)
The increased rate of net catabolism in sepsis can result from a rise in protein degradation, or a decrease in protein synthesis, or both. The relative importance of protein synthesis versus degradation in the induction of the negative nitrogen balance during sepsis is unresolved. The increased net catabolism after trauma uncomplicated by sepsis results from an inhibition of protein synthesis rather than a change in protein degradation.[54] [58] [59] In contrast, sepsis accelerates muscle proteolysis.[56] [60]
The rate of protein synthesis in mixed hind limb muscles is not altered in nonseptic inflammation, whereas sepsis produces a 50% inhibition of protein synthesis.[61] [62] This reduction did not result from a decreased RNA content. Instead, the translational efficiency, expressed as protein synthesis/RNA, was significantly reduced by 50% in skeletal muscle of septic animals compared with controls. The effect of sepsis to lower protein synthesis was further examined by use of individual muscles containing different fiber types.[61] Gastrocnemius and psoas muscles were chosen to represent fast-twitch muscles, whereas soleus and heart muscles represented slow-twitch muscles. Sepsis caused a decrease in the muscle weight of all four muscles. The protein concentration and the protein synthetic rate in fast-twitch muscle were reduced by sepsis, whereas neither of these parameters was affected in slow-twitch or heart muscles. The sepsis-induced inhibition of protein synthesis in fast-twitch muscles resulted from a restraint in peptide-chain initiation because sepsis caused a 1.6-fold increase in free ribosomal subunits.[55] [61]
The net catabolic state in skeletal muscle is characterized by changes in the intracellular amino acid concentrations. Postoperatively, the concentrations of branched-chain amino acids (i.e., phenylalanine, tyrosine, and methionine) increase compared with preoperative values. Although the basic amino acids show small reductions, the overriding feature of injury is a depletion of glutamine concentrations.
The concentration of glutamine in human muscle is extremely high (20 mM) and is equivalent to 715 g of nitrogen in the adult. Glutamine, which represents as much as 65% of the total free amino acids in muscle, decreases by up to 50% immediately after trauma.[49] [63] [64] The muscle glutamine constitutes a labile pool that could influence changes in whole-body nitrogen balance. This decrease in glutamine cannot be rectified by administration of total parental nutrition (TPN) support because most TPN solutions do not contain glutamine. Glutamine is unique among the amino acids because muscle glutamine concentrations are inversely related to rates of
Glutamine arises from de novo synthesis and from release from protein stores. Glutamine is synthesized from α-ketoglutarate, a citric acid cycle intermediate, by glutamate and transamination reactions. Glutamine synthase, a multienzyme complex, catalyzes this reaction, and its concentration is particularly high in skeletal muscle and kidney. After amination of glutamate to glutamine, glutamine is released into the bloodstream. It is the most important carrier for removal of nitrogen from peripheral tissues to the splanchnic area.
The role of this labile pool of glutamine in skeletal muscle has not been fully elucidated. Possible functions of increased glutamine release by muscle are to serve as a labile store of fuel for intestinal cells and lymphocytes and a source of ammonia for acid-base imbalances. As a fuel, cells use glutamine for two major processes. First, it can be used as an energy source. Cells of the small intestine use glutamine for oxidative energy metabolism. The output of ammonia, protein, alanine, glutamate, and citrulline from gut cells depends on the supply of glutamine to the gut. Second, glutamine is a donor of nitrogen in de novo purine and pyridine biosynthesis and is therefore essential for cellular proliferation. An intraorgan relationship between glutamine release by skeletal muscle and use by splanchnic organs exists.
Decreased skeletal muscle glutamine concentrations postoperatively can result from decreased glutamine synthase activity, decreased release of glutamine from protein stores, increased release of glutamine from muscle through accelerated transport activity, or increased breakdown of glutamine by glutaminase. It is unlikely that increased glutaminase activity is responsible for the decrease in muscle glutamine in sepsis because functional hepatectomy causes a significant accumulation of glutamine in the blood.[51] It argues against decreased glutamine synthase activity. Glutamine synthase activity in skeletal muscle traumatized by 1-carrageenan is markedly increased, not decreased.[66]
Inhibition of glutamine synthase with methionine sulfoximine attenuates glutamine release by 50%, suggesting that release of glutamine from protein stores is also an important mechanism in glutamine production.[52] Despite increased production of glutamine by de novo synthesis and increased proteolysis, the skeletal muscle glutamine concentrations are reduced. An increase in glutamine transport must also occur.
To determine whether decreased glutamine is critical in post-traumatic skeletal muscle protein catabolism, glutamine has been administered as part of TPN. In this regard, the postoperative efflux of amino acids from dog hind limb was reduced by infusing an amino acid mixture supplemented with glutamine.[67] However, commercially available amino acid supplements do not contain glutamine because it is considered a nonessential amino acid and is unstable in aqueous solutions. Consequently, it must be added immediately before use. A more convenient method of supplying glutamine in TPN solutions is to use stable dipeptides, such as alanyl-glutamine and glycyl-glutamine. Both dipeptides are hydrolyzed in the body, releasing glutamine. Administration of these dipeptides as part of TPN counteracts the decrease in muscle glutamine and improves nitrogen balance in the immediate postoperative period[68] ( Fig. 77-17 ).
In addition to provision of dipeptides, plasma and skeletal muscle glutamine concentrations can be augmented by the provision of ornithine-α-ketoglutarate or α-ketoglutarate. Supplementation of TPN with either of these compounds maintains nitrogen balance and decreases cumulative urea excretion after abdominal surgery.[69] Attention has been focused on the ability of ornithine α-ketoglutarate to selectively preserve the mucosal barrier and thereby prevent bacterial translocation across the gut. Ornithine α-ketoglutarate limits bacterial dissemination and metabolic changes after injury in rats and may be useful in prevention of gut-derived sepsis in critically ill patients. The rate of protein synthesis is maintained, and the characteristic increase in several essential amino acids in skeletal muscle is also attenuated.
The stimulus for enhanced muscle catabolism during sepsis is unknown. A certain amount of catabolic response in trauma patients can be attributed to direct injury to the muscle. However, the enhanced catabolism in the septic patient occurs without any overt signs of direct tissue trauma. The amount of catabolism observed in septic patients is far in excess of what would be expected simply because these patients are bedridden and inactive.
In burn patients, cortisol is a major determinant of the catabolic response.[70] The catabolic effects of cortisol have been confirmed in healthy volunteers subjected to cortisol infusions to levels seen in burn patients.
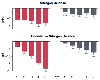 Figure 77-17
Day-to-day and cumulative nitrogen balance in patients
receiving 1-alanyl-1-glutamine dipeptide-supplemented (filled
columns) or conventional-amino-acid total parenteral nutrition (TPN) (open
columns). These results illustrate a beneficial effect for increasing
glutamine concentration in maintaining nitrogen balance. ***P
<.001; **P < .01. (Adapted
from Stehle P, Zander J, Mertes N, et al: Effect of parenteral glutamine peptide
supplements on muscle glutamine loss and nitrogen balance after major surgery. Lancet
1:231, 1989.)
Figure 77-17
Day-to-day and cumulative nitrogen balance in patients
receiving 1-alanyl-1-glutamine dipeptide-supplemented (filled
columns) or conventional-amino-acid total parenteral nutrition (TPN) (open
columns). These results illustrate a beneficial effect for increasing
glutamine concentration in maintaining nitrogen balance. ***P
<.001; **P < .01. (Adapted
from Stehle P, Zander J, Mertes N, et al: Effect of parenteral glutamine peptide
supplements on muscle glutamine loss and nitrogen balance after major surgery. Lancet
1:231, 1989.)
Besides cortisol, epinephrine concentrations are elevated in sepsis. In contrast to cortisol, the catecholamines may not have as dramatic an effect on protein turnover.[72] Isoproterenol does not alter protein synthesis but does reduce protein degradation by 20%. The decreased proteolysis is sufficient to prevent the release of alanine, threonine, phenylalanine, tyrosine, lysine, arginine, leucine, and valine.
Insulin conserves muscle protein by stimulating protein synthesis and by inhibiting protein degradation.[73] [74] However, in sepsis, the breakdown of muscle protein, as measured by 3-methylhistidine release, is increased despite increased insulin concentrations.[56] These observations also suggest that additional factors resulting from the septic episode accelerate muscle catabolism or that injury itself renders the muscle resistant to insulin action.
Aside from insulin, GH promotes nitrogen retention and improves nitrogen balance in a variety of catabolic conditions.[75] Septic patients have inappropriately high GH concentrations indicative of a severe GH resistance. GH has a reduced effectiveness to limit protein catabolism in septic patients.[76] [77] Moreover, administration of GH is associated with an increased morbidity and mortality in critically ill patients.[78] GH administration may have limited usefulness in the treatment of septic patients.
Insulin-like growth factor-1 (IGF-1) is believed to mediate the anabolic action of GH in muscle. Consequently, IGF-1 may be of more importance than GH in improving nitrogen balance in skeletal muscle during sepsis. In myotubes or myoblasts from the L8 or L6 cell lines in culture, IGF-1 is a more potent stimulator of protein synthesis than insulin.[79] [80] Intravenous infusion of IGF-1 directly increases protein synthesis in skeletal muscle provided adequate substrate supply is also present.[81] [82] Systemically infused IGF-1 stimulates weight gain and protein synthesis in normal rats; increases lean body mass during starvation, acquired immunodeficiency syndrome (AIDS), or diabetes; attenuates protein loss during glucocorticoid-induced cachetic states and in pediatric burn patients; and improves nitrogen balance in endotoxin.[83] [84] [85] [86] [87] [88] [89] [90] [91]
There is a positive linear correlation between the IGF-1 content in muscle and rate of protein synthesis.[92] The strong correlation between IGF-1 and protein synthesis in gastrocnemius does not prove cause and effect; however, such a relationship is consistent with in vitro studies demonstrating an ability of IGF-1 to stimulate muscle protein synthesis. IGF-1 stimulates skeletal muscle protein synthesis during sepsis or severe acute peritonitis.[93] [94] [95] Based on the responsiveness of skeletal muscle to IGF-1 in vitro and the decreased plasma concentrations of IGF-1 during sepsis, alterations in the bioavailability of IGF-1 would be expected to have profound effects on rates of protein synthesis in skeletal muscles. Enhancing the bioavailability of IGF-1 through administration of IGFF-IGFBP-3 complex stimulates the rate of protein synthesis in gastrocnemius by enhancing the translational efficiency in vivo.[96]
The IGF-1 system is complex and multifactorial being composed of the hormone and at least six different IGF-binding proteins (IGFBPs). IGFBPs carry the IGF-1 in the blood and modulate its bioavailability, thereby inhibiting or potentiating the interaction of IGF-1 with its receptor. In this regard, a common feature of catabolic conditions, including sepsis, is an elevation in the plasma IGFBP-1 and IGFBP-2 concentration.[97] [98] [99] An increase in the plasma IGFBP-1 concentration would be expected to sequester IGF-1, lowering the effective IGF-1 concentration in plasma of septic rats. Treatment of incubated muscles with anti-IGF-1 antibodies reduced rates of protein synthesis, indicating that limiting IGF-1 bioavailability is associated with decreased rates of protein synthesis.[100] Unlike these two binding proteins, IGFBP-3 is reduced in critically ill humans, but similar changes in plasma IGFBP-3 are not observed in rodent models of sepsis. Infusion of interleukin-1 Ra (IL-1Ra) completely prevented the rise in IGFBP-1 levels but had no effect on plasma IGFBP-2 or IGFBP-3 concentrations.[101]
The factors responsible for the septic-induced metabolic alterations are unknown. Although the cause of potential mediators may be multifactorial, a frequent common denominator of organ dysfunction and organ failure is alterations in the release of cytokines, particularly by the macrophage. Tumor necrosis factor (TNF), IL-1, and IL-6 play a pivotal role in mediating some of the metabolic responses to sepsis. Cytokines elicit various and often overlapping effects designed to protect the host in response to an inflammatory or bacterial insult. As a mediator of the septic process, TNF triggers neutrophils to degranulate, lymphocytes to divide, and other cytokines (including IL-1 and IL-6) to be released. The natural induction of cytokines during inflammation is beneficial, but overproduction, as occurs in sepsis, is detrimental to the host. The proinflammatory cytokines (i.e., TNF, IL-1, and IL-6) may play a role in mediating the effects of sepsis on skeletal muscle protein metabolism. One approach to understanding the role for these cytokines in mediating the inhibition in skeletal muscle protein synthesis is to modify their release or biologic action, or both, during sepsis. Administration of phosphodiesterase inhibitors, such as pentoxifylline (PTX) or amrinone, decreases the serum TNF concentration of endotoxin-treated mice, rats, and humans and after induction of sepsis in rats.[102] Suppression of TNF secretion by PTX partially prevented the inhibition of protein synthesis and stimulation of protein degradation during sepsis. PTX treatment improves the ability of insulin to improve skeletal muscle protein balance during the anorexic (2 days after infection) and hypermetabolic (6 days after infection) phases after injection of bacteria.[102] Cytokines may influence skeletal muscle protein metabolism during sepsis by directly inhibiting the protein synthetic machinery and indirectly by promoting an insulin resistance.
The biologic activity of TNF is modulated in vivo by the proteolytic shedding of the extracellular domain of the p55 and p75 TNF receptors. An increase in soluble TNF receptors in the bloodstream neutralizes circulating TNF, thereby lowering the biologically active concentration of TNF in the plasma. TNF binding protein is a dimeric, polyethylene glycol-linked form of the human p55 soluble
IL-1 is a logical choice as a mediator of sepsis-induced abnormalities in protein synthesis. Infusion of IL-1 significantly decreased muscle weight, protein content, and the rate of protein synthesis in gastrocnemius (i.e., fast-twitch muscle), but it had little or no effect on these parameters in soleus (i.e., slow-twitch muscle) compared with controls.[104] The changes in protein metabolism with chronic IL-1 infusion resemble those observed in chronic abdominal sepsis. These results provide additional evidence that IL-1-induced alterations in protein synthesis may contribute to changes in skeletal muscle observed in catabolic illness. Blockage of IL-1 action with a specific IL-1 receptor antagonist prevents the fall in muscle mass by maintaining rates of protein synthesis.[105] Infusion of IL-1Ra in septic patients elevates the plasma concentration of eleven amino acids, and total amino acid concentration was increased by 50% within 70 hours and was associated with a lower nitrogen excretion.[106] Concentrations of several amino acids, including leucine, were increased up to twofold by infusion of IL-1Ra, which therefore may be a useful adjunct to promote muscle accretion during sepsis.
In addition to the role of leucine in de novo alanine synthesis, the branched-chain amino acids are of special importance because some reports have suggested that administration of branched-chain amino acids is beneficial in septic patients, improving the survival in surgical patients with multiple organ failure. [107] [108] [109] Administration of branched-chain amino acids as part of the nutritional support in traumatized or septic animals results in decreased weight loss, reduced negative nitrogen balance, decreased protein catabolism, and increased protein synthesis. In surgical patients, nitrogen retention was greater in those patients receiving TPN with 45% of the amino acids as branched-chain amino acids than those patients receiving TPN with 24% of the amino acids as branched-chain amino acids. One hypothesis for their efficacy is that they provide a fuel for skeletal muscle and liver energy metabolism at a time when glucose oxidative metabolism may be inhibited.
Branched-chain amino acids are among the few amino acids oxidized in muscle tissues. In healthy animals and humans, there appears to be a differential pattern of branched-chain amino acid metabolism in muscle and liver. In muscle tissues, the aminotransferase activity is high, and the oxidative decarboxylation of the corresponding α-ketoacid is rate limiting for leucine oxidation. Only about 50% of the leucine undergoing transamination is oxidized. The corresponding α-ketoacids are released into the blood. The concentration of all three α-ketoacids of branched-chain amino acids in human plasma is approximately 0.1 to 0.04 mM. In contrast to muscle, in hepatic tissue, the activity of the branched-chain dehydrogenase is higher than that of the transaminase, and α-ketoacids are readily oxidized. The reason for differential pattern may lie in the differential regulation of the branched-chain amino acid dehydrogenase in muscle and liver. The branched-chain α-ketoacid dehydrogenase is also regulated by a phosphorylation/dephosphorylation cycle, analogous to the PDH complex.[110] The branched-chain α-ketoacid dehydrogenase is inactivated in muscle tissue through phosphorylation but is protected in some way from inactivation in liver. This affords a mechanism whereby α-ketoacids released by extrahepatic tissues are taken up and used by the liver with the possible formation of glucose and ketone bodies.
The absolute contribution of branched-chain amino acids to the overall energy metabolism (i.e., adenosine triphosphate generation) is still debatable. In the absence of any other exogenous substrate, leucine oxidation in cardiac muscle could only account for about 5% of the total oxygen consumption, even at supraphysiologic levels of leucine.[111] Leucine could not provide sufficient energy to maintain normal cardiac function. Hence, it is doubtful that leucine represents a significant energy fuel.
Alternatively, the branched-chain amino acids (leucine in particular) are unique in that they stimulate protein synthesis.[112] [113] The stimulatory effect of branched-chain amino acids on protein synthesis was associated with a facilitated efficiency of translation. However, the response is not as great as that produced by insulin. The protein-sparing effects of branched-chain amino acids are more likely a result of modulation of protein metabolism.
Although net proteolysis occurs in skeletal muscle in sepsis, protein synthesis in liver after trauma or infection is increased (see Chapter 19 ). The increased rates of protein synthesis coincide with enhanced hepatic uptake (or clearance) of amino acids. The increased hepatic protein synthesis has been suggested to be caused by or dependent on an increased supply of amino acids from the peripheral tissues. Liver is unique in that it synthesizes proteins for its own use (i.e., structural proteins essential for normal cell function), as well as proteins that are secreted into the blood (i.e., secretory proteins such as albumin). The synthesis of secreted and nonsecreted proteins was stimulated in inflammation and sepsis[114] ( Table 77-4 ). There was a preferential stimulation of proteins destined for secretion, the acute-phase proteins.
Distinct and coordinated changes in the concentration of individual plasma proteins, called acute-phase proteins, occur within 24 hours after injury. [115] The acute-phase proteins maximize immune responsiveness to the foreign body and repair to damaged tissues. These functions include complement activation and opsonization needed for bacterial killing (i.e., C-reactive protein), coagulation, surface structure, and support lattice formations needed for leukocyte entrapment of foreign material (i.e., fibrinogen), superoxide scavenging (i.e., ceruloplasmin), and inactivation of excess proteases needed to prevent damage to viable cells
| Measurement | Control | Sterile Abscess | Septic Abscess |
|---|---|---|---|
| Total secreted protein production (mg/g/hr) | 1.04 ± 0.07 | 1.81 ± 0.17 ‡ | 1.48 ± 0.07 ‡ |
| Synthesis of nonexported protein (mg/g/hr) | 1.60 ± 0.08 | 2.14 ± 0.14 § | 1.95 ± 0.10 ¶ |
| Synthesis of total liver protein (mg/g/hr) * | 2.56 ± 0.13 | 3.94 ± 0.29 ‡ | 3.37 ± 0.10 ‡ |
| Percent of total liver protein secreted † | 39 ± 2 | 47 ± 2 ¶ | 47 ± 1 ¶ |
| From Vary TC, Kimball SR: Regulation of hepatic protein synthesis in chronic inflammation and sepsis. Am J Physiol 262:C445, 1992. | |||
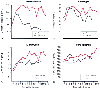 Figure 77-18
Hepatic acute-phase (AP) protein response after trauma
in 10 septic and 16 nonseptic patients. Mean values are provided for C-reactive
protein (CRP) (A), fibrinogen (B),
α1
-antitrypsin (C), and ceruloplasmin
(D). Patients with sepsis developing after trauma
are compared with patients with nonseptic post-trauma courses. Notice the early
rise of CRP and α1
-antitrypsin levels in patients who became clinically
septic at 5 to 7 days. The fibrinogen response and ceruloplasmin response tend to
be increased in sepsis, although only after the fully developed septic clinical picture
is demonstrated. (Adapted from Sganga G, Siegel JH, Brown G, et al: Reprioritization
of hepatic plasma protein release in trauma and sepsis. Arch Surg 120:187, 1985.)
Figure 77-18
Hepatic acute-phase (AP) protein response after trauma
in 10 septic and 16 nonseptic patients. Mean values are provided for C-reactive
protein (CRP) (A), fibrinogen (B),
α1
-antitrypsin (C), and ceruloplasmin
(D). Patients with sepsis developing after trauma
are compared with patients with nonseptic post-trauma courses. Notice the early
rise of CRP and α1
-antitrypsin levels in patients who became clinically
septic at 5 to 7 days. The fibrinogen response and ceruloplasmin response tend to
be increased in sepsis, although only after the fully developed septic clinical picture
is demonstrated. (Adapted from Sganga G, Siegel JH, Brown G, et al: Reprioritization
of hepatic plasma protein release in trauma and sepsis. Arch Surg 120:187, 1985.)
Because the liver is responsible for the synthesis of these acute-phase proteins, hepatic protein synthesis is accelerated by a wide variety of acute inflammatory insults. Despite the accelerated rate of hepatic protein synthesis during inflammation and sepsis, the mechanisms responsible for this stimulation have not been fully elucidated. The synthesis of specific proteins is probably regulated transcriptionally during inflammation. For example, messenger RNA concentrations for the acute-phase proteins are elevated,[116] [117] [118] whereas albumin messenger RNA is decreased. [119] However, increased messenger RNA concentrations may not wholly account for the overall increased rates of protein synthesis. Instead, the translation phase of the protein synthetic pathway is also increased.[114] IL-6 appears to be responsible for the changes in acute-phase messenger RNA levels in trauma and sepsis.[120]
The metabolic course of the traumatized or septic patient shows that fatty acids become the preferred fuel for oxidative metabolism. The same dependence on fatty acid metabolism is observed under conditions of starvation and diabetes, in which the conservation of glucose carbon becomes pathologic ( Fig. 77-19 ).
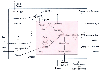 Figure 77-19
Septic alteration of metabolic regulation of hepatic
fatty acid oxidation, ketogenesis, fatty acid synthesis, and carbohydrate metabolism.
Malonyl CoA (dotted line) derived from citrate and
cytosolic acetyl CoA inhibits carnitine:acyl CoA transferase I. (carnitine:acyl
CoA transferases [I and II] are bound to inner mitochondrial membrane in vivo, but
for clarity, the reactions are shown away from the membrane.) Hormones (i.e., insulin
or glucagon) and small and large septic abscesses can increase (+) or inhibit (-)
particular synthetic pathways or oxidation of specific substrates. CAT-1, carnitine:acyl
CoA transferase I; CE, cholesterol esters; CoA, coenzyme A; FA-CoA, long-chain fatty
acyl CoA; FACarn, long-chain fatty acyl carnitine; FFA, long-chain fatty acids; HMG-CoA,
β-hydroxy-β-methyl glutaryl-CoA; LACT, lactate; Mito, mitochondria; OAA,
oxaloacetate; PDH, pyruvate dehydrogenase complex; PL, phospholipids; PYR, pyruvate;
TCA, tricarboxylic cycle; TG, triglycerides; II, carnitine:acyl transferase II.
(Adapted from Vary TC, Siegel JH, Nakatani T, et al: A biochemical basis
for depressed ketogenesis in sepsis. J Trauma 26:419, 1986.)
Figure 77-19
Septic alteration of metabolic regulation of hepatic
fatty acid oxidation, ketogenesis, fatty acid synthesis, and carbohydrate metabolism.
Malonyl CoA (dotted line) derived from citrate and
cytosolic acetyl CoA inhibits carnitine:acyl CoA transferase I. (carnitine:acyl
CoA transferases [I and II] are bound to inner mitochondrial membrane in vivo, but
for clarity, the reactions are shown away from the membrane.) Hormones (i.e., insulin
or glucagon) and small and large septic abscesses can increase (+) or inhibit (-)
particular synthetic pathways or oxidation of specific substrates. CAT-1, carnitine:acyl
CoA transferase I; CE, cholesterol esters; CoA, coenzyme A; FA-CoA, long-chain fatty
acyl CoA; FACarn, long-chain fatty acyl carnitine; FFA, long-chain fatty acids; HMG-CoA,
β-hydroxy-β-methyl glutaryl-CoA; LACT, lactate; Mito, mitochondria; OAA,
oxaloacetate; PDH, pyruvate dehydrogenase complex; PL, phospholipids; PYR, pyruvate;
TCA, tricarboxylic cycle; TG, triglycerides; II, carnitine:acyl transferase II.
(Adapted from Vary TC, Siegel JH, Nakatani T, et al: A biochemical basis
for depressed ketogenesis in sepsis. J Trauma 26:419, 1986.)
The release of fatty acids from adipose tissue in sepsis is variable, with some reports of an increased release and other reports of a decreased release. Despite unaltered arterial fatty acid concentrations, fatty acids are continually released into the bloodstream and are delivered to the liver. After removal from plasma, fatty acids may undergo oxidation for energy and ketone body production, or esterification to triglycerides. Increased hepatic triglyceride levels are a characteristic feature of sepsis, giving rise to the histologic observations of increased lipid droplets in liver tissue at autopsy. The increased triglyceride levels may partly result from re-esterification. This increased re-esterification may be responsible in part for the increased secretion of triglycerides as very-low-density lipoproteins. Administration of monokines (i.e., TNF or IL-1) increases hepatic lipogenesis in fed rats but not in fasted rats.[121] [122]
Although ketogenesis may be inhibited during sepsis, lipogenesis appears to be accelerated. In septic patients receiving only glucose in excess of 800 cal/m2 body surface area, the respiratory quotient (RQ) rises above 1.0, a value that is indicative of net lipogenesis.[44] [45] The capacity to
The potential site of hepatic fatty acid oxidation regulation, and therefore of ketone body formation, is carnitine: acyl transferase-1. This enzyme is competitively inhibited by malonyl CoA, resulting in decreased formation of long-chain fatty acyl carnitine. Malonyl CoA is the first committed intermediate in the conversion of glucose carbon into fat, and its concentration is known to fluctuate in parallel with the rate of fatty acid synthesis. The physiologic role of malonyl CoA is to provide unidirectional flow of glucose carbon to fatty acid synthesis by preventing the futile oxidation of newly synthesized fatty acids by the carnitine:acyl transferase system to β oxidation. The malonyl CoA concentration is elevated twofold in livers from septic animals.[123] The rise in malonyl CoA concentration is in the range reported for maximal inhibition of carnitine:acyl transferase-1. The increased malonyl CoA concentrations would be expected to inhibit the carnitine:acyl transferase-1 system during sepsis.
Progressive sepsis increases plasma triglycerides as the septic process worsens. Besides an increase in hepatic triglyceride synthesis, the peripheral triglyceride disposal mechanisms may be impaired. Lipoprotein lipase activity, the enzyme responsible for clearance of plasma triglycerides, is reduced in adipose tissue and muscle from septic animals. Concomitant with the lowered lipoprotein lipase activity, plasma triglyceride concentrations are increased. It appears that a monokine or several monokines acting synergistically act to downregulate lipoprotein lipase activity.
The normal response of the liver to increased delivery of fatty acids is the synthesis of ketone bodies (i.e., 3-L-hydroxybutyrate and acetoacetate). Reversal of the insulin-to-glucagon ratio enhances the ketogenic capacity of the liver. In sepsis, there is a rise in the glucagon-to-insulin ratio, but the plasma ketone body concentrations are lower than expected given the hormonal environment, and septic animals and patients undergoing a total fast do not demonstrate ketonemia. The lack of elevated plasma ketones does not indicate a lack of fatty acid oxidation, because these patients have an RQ of 0.75, which is indicative of fat oxidation. Sepsis appears to induce changes in hepatic fatty acid metabolism that prevent or reverse maximal rates of ketogenesis.
The failure of hepatic tissue to enhance ketogenesis may be important to the clinical outcome of the septic episode, because survival depends on normal liver function. A decrease in the circulating ketone body concentrations would be expected to increase the dependence of peripheral tissues on alternative substrates for energy production when cellular metabolism is accelerated by sepsis. The failure of similar increases in plasma ketone bodies in nondiabetic septic patients may be an additional factor in the markedly increased rates of proteolysis in septic patients. Of considerable interest is the observation that enhanced muscle catabolism in response to injury is not observed if the blood concentration of ketone bodies is increased. However, it would be impractical to simply supply ketone bodies as nutritional support. To replace 5% glucose, the patient would require 125 g of ketones/day. That amount of ketone bodies presents few problems, because ketone bodies are readily soluble in saline. The problem is that accompanying the ketone body infusion would be approximately 1200 mEq of a positive ion (Na+ ). Such a salt load on the kidneys is certainly not advised in healthy persons and is probably devastating in the septic patient, who may have complicating renal dysfunction.
The effects of sepsis on glucose production in liver are well characterized, but very little information is available concerning the effect of sepsis on peripheral glucose use. Abnormal glucose tolerance tests are commonly observed after traumatic injury, burn, shock, or sepsis despite normal or accentuated insulin secretion. Despite the responsiveness of the pancreatic β-cells to secrete insulin in response to a glucose load, glucose intolerance and hyperglycemia persist, suggesting that certain target organs in the injured or septic patient are relatively insensitive to the effects of circulating insulin. Because glucose consumption by central and peripheral nervous tissue, renal medulla, bone marrow, erythrocytes, and leukocytes is not insulin sensitive, the primary sites of insulin resistance are in peripheral tissues, particularly skeletal muscle and adipose tissue, where insulin stimulates glucose uptake. Because only a small percentage (1%) of the glucose load is taken up by adipose tissue, the major effect of insulin appears to be in muscle tissue.
In the case of sepsis, insulin resistance is manifested by an abnormal glucose tolerance test result or by an elevated plasma glucose concentration for a given insulin concentration. This insulin resistance could occur by alterations at one of three levels: before the interaction of insulin with the receptor, at the receptor level, or at steps distal to the insulin-receptor interaction (i.e., cellular metabolism). Insulin concentration is the same or increased in sepsis, and no anti-insulin antibodies have been detected. At the receptor level, the sensitivity of the receptor to circulating insulin appears to be normal.[124] It appears that insulin resistance in sepsis may be related to an intracellular defect in glucose metabolism.
Induction of sepsis in rats also causes an inhibition of protein synthesis in skeletal muscle that is resistant to the anabolic actions of insulin. [125] [126] To gain a better understanding of the underlying reason for this lack of response, sepsis-induced alterations in insulin signaling to regulatory components of mRNA translation were established.[126] The insulin-induced hyperphosphorylation response of the translation repressor protein, 4E-BP1, and of the 70-kd S6 kinase, S6K1, two targets of insulin action on mRNA translation, was unimpaired in gastrocnemius of septic rats. Hyperphosphorylation of 4E-BP1 in response to insulin resulted in its dissociation from the inactive eIF4E·4E-BP1 complex in control and septic rats. Assembly of the active eIF4F complex as assessed by the association eIF4E with eIF4G did not follow the pattern
|
|
|
|
|
|
|
|
|
|
|
|
|