|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The relatively academic controversy about volatile anesthetic-induced coronary vasodilation and coronary steal has detracted from substantial experimental evidence indicating that volatile agents exert important protective effects during myocardial ischemia and reperfusion injury. Halothane attenuates ST-segment changes caused by brief coronary artery occlusion[297] [298] and decreases ST-segment elevation to a greater extent than propranolol and sodium nitroprusside despite producing similar hemodynamic effects.[298] Halothane and isoflurane reduce myocardial infarct size after coronary artery ligation. [55] [56] [299] [300] [301] Enflurane decreases cardiac lactate production in the presence of a critical coronary artery stenosis during artificial control of perfusion pressure.[57] Isoflurane and desflurane produce beneficial actions on LV diastolic mechanics during acute regional myocardial ischemia.[61] Halothane, enflurane, and isoflurane decrease myocardial reperfusion injury and improve functional recovery after global ischemia in isolated hearts.[302] [303] [304] [305] Volatile agents also enhance systolic functional recovery of postischemic-reperfused ("stunned") myocardium when these agents are administered before and during,[58] [60] but not after,[306] [307] brief periods of myocardial ischemia in vivo ( Fig. 7-18 ). These effects are accompanied by
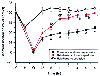 Figure 7-18
Segment shortening data (expressed as a percentage of
the control value) during a 15-minute coronary artery occlusion (O) and at various
times after reperfusion in the conscious (C) and halothane-anesthetized (H) states.
Comparisons are made at various time points with animals anesthetized with halothane
for 2.25 hours and allowed to emerge from anesthesia over a 5-hour period but not
undergoing coronary artery occlusion and reperfusion. (Adapted from Warltier
DC, Al-Wathiqui MH, Kampine JP, Schmeling WT: Recovery of contractile function of
stunned myocardium in chronically instrumented dogs is enhanced by halothane or isoflurane.
Anesthesiology 69:552–565, 1988.)
Figure 7-18
Segment shortening data (expressed as a percentage of
the control value) during a 15-minute coronary artery occlusion (O) and at various
times after reperfusion in the conscious (C) and halothane-anesthetized (H) states.
Comparisons are made at various time points with animals anesthetized with halothane
for 2.25 hours and allowed to emerge from anesthesia over a 5-hour period but not
undergoing coronary artery occlusion and reperfusion. (Adapted from Warltier
DC, Al-Wathiqui MH, Kampine JP, Schmeling WT: Recovery of contractile function of
stunned myocardium in chronically instrumented dogs is enhanced by halothane or isoflurane.
Anesthesiology 69:552–565, 1988.)
Volatile anesthetics may also produce beneficial effects on blood flow to ischemic myocardium. Decreases in collateral blood flow after coronary artery occlusion are less pronounced than declines in flow to normal myocardium in the presence of halothane.[309] The ratio of myocardial oxygen delivery to MVO2 is also increased in collateral-dependent myocardium during halothane anesthesia.[309] Evidence indicates that sevoflurane actually increases blood flow to collateral-dependent myocardium when arterial pressure is maintained at conscious values.[294] Halothane may inhibit platelet thrombi formation through increases in platelet cAMP concentration and decrease cyclical variations in coronary blood flow associated with a critical coronary artery stenosis.[310] Volatile anesthetics can attenuate the adhesion of neutrophils and platelets in the coronary vasculature after ischemia and reperfusion.[311]
The mechanisms responsible for volatile anesthetic-induced protection during myocardial ischemia and reperfusion are incompletely understood. The beneficial effects of volatile anesthetics may be attributed to a favorable reduction in myocardial oxygen demand required for active contraction with concomitant preservation of energy-dependent vital cellular processes because these anesthetics cause direct negative inotropic, lusitropic, and chronotropic effects and decrease LV afterload. However, halothane also exerts protective effects during complete functional arrest induced by cardioplegia, [59] indicating that preferential alterations in myocardial oxygen supply-demand relationships are not solely responsible for the anti-ischemic actions of this anesthetic. Halothane, enflurane, and isoflurane may significantly lower excessive intracellular Ca2+ during reperfusion through a direct decline in the net transsarcolemmal Ca2+ transient resulting from partially inhibited Ca2+ channel activity[69] [71] [72] [73] or an indirect reduction of oxygen-derived free radical formation.[308]
Evidence indicates that the protective effects of isoflurane during ischemic injury are mediated by activation of KATP channels.[280] KATP channels are heteromultimeric complexes consisting of an inward-rectifying K+ channel (Kir ) and a sulfonylurea receptor (SUR).[312] Pharmacologically distinct KATP channels have been identified in cardiac sarcolemmal[313] and mitochondrial[314] membranes. It was originally proposed that opening of sarcolemmal KATP channels protects ischemic myocardium by shortening action potential duration and preventing intracellular Ca2+ overload.[313] However, subsequent studies indicated that the beneficial actions of KATP channel opening occurred independent of the action potential duration.[315] [316] Most experimental findings suggest that mitochondrial KATP channels are the primary mediators of ischemia-induced[317] and anesthetic-induced preconditioning (IPC and APC, respectively). Preservation of mitochondrial bioenergetic function appears to be vital for cellular protection against myocardial ischemia.[318] [319] [320] [321] Mitochondrial KATP channel openers maintain intracellular Ca2+ homeostasis and inhibit mitochondrial Ca2+ overload.[320] [321] These actions enhance myocyte survival by preventing tissue necrosis or apoptosis.[322] Mitochondrial KATP channel activation inhibits apoptosis in rat ventricular myocytes [323] by attenuating oxidant stress during reperfusion. [324] Alteration of the mitochondrial oxidation-reduction state by KATP channel activation may also promote cellular protection. [325] Experiments conducted in isolated cardiac mitochondria[320] indicate that membrane depolarization, matrix swelling, and uncoupling of ATP synthesis occur as a result of increased oxygen consumption, are associated with the opening of mitochondrial KATP channels, and may mediate cellular viability during IPC.[325] Opening of mitochondrial KATP channels depolarizes the inner mitochondrial membrane and causes a transient swelling of the mitochondrial matrix,[326] resulting from a shift in the ionic balance.[327] Mitochondrial KATP channel opening initially reduces ATP production as a result of membrane depolarization[320] but subsequently stimulates a compensatory increase in respiration that optimizes the efficiency of oxidative phosphorylation, in part by regulating energy-dependent matrix volume. [328] A moderate disturbance of mitochondrial homeostasis may promote myocardial tolerance to ischemic stress by altering energetic systems to reduce Ca2+ overload, prevent the activation of necrotic or apoptotic pathways, or attenuate oxidant stress.
The endogenous mechanisms involved in APC are strikingly similar to those implicated in IPC. It is hypothesized that a trigger (e.g., brief ischemic episode, exposure to a volatile anesthetic) initiates a cascade of signaling events leading to activation of an end-effector that is responsible for resistance to injury. KATP channel activation has been implicated as the end-effector in this protective scheme during APC. Administration of isoflurane and sevoflurane preserved myocyte viability using a cellular model of ischemia compared with cells that were not exposed to a volatile agent.[329] This protective effect was abolished by the selective mitochondrial KATP channel antagonist 5-hydroxydecanoate (5-HD) but not the selective sarcolemmal KATP channel antagonist HMR-1098.[329] The nonselective KATP channel blocker glyburide attenuated the recovery of contractile function produced by isoflurane in stunned myocardium ( Fig. 7-19 ).[279] [330] Isoflurane-induced reductions in canine myocardial infarct size[56] ( Fig. 7-20 ) and the ATP-sparing effects of this agent[331] are also abolished by glyburide. 5-HD inhibits preconditioning by isoflurane in rats,[332] rabbits,[333] and isolated human atria.[334] [335] HMR-1098 and 5-HD abolished the protective effects of desflurane in dogs,[336] supporting a role for sarcolemmal and mitochondrial KATP channels in APC. The latter data contrast to some degree with findings that implicate mitochondrial KATP channels as the predominant mediators of IPC.[317]
Results of in vitro experiments also strongly suggest that volatile anesthetics affect KATP channel function. Isoflurane stimulated outward K+ current through sarcolemmal KATP channels in isolated ventricular myocytes during patch-clamping.[337] [338] Volatile anesthetics also reduced sarcolemmal KATP channel sensitivity to inhibition by ATP, thereby increasing the open-state probability.[339]
 Figure 7-19
Percent segment shortening (%SS) in the intermittently
ischemic and reperfused left anterior descending coronary artery (LAD) region. The
%SS decreases significantly (P < .05) from baseline
during each 5-minute LAD occlusion and reperfusion in all groups. Significant decreases
in %SS during each 5-minute reperfusion and throughout 180 minutes of final reperfusion
are observed in dogs pretreated with glyburide, a KATP
channel antagonist,
in the presence or absence of isoflurane. The %SS recovers to baseline values after
reperfusion in dogs receiving isoflurane alone. †, Significantly (P
< .05) different from dogs pretreated with drug vehicle; §, significantly
(P < .05) different from dogs pretreated with
glyburide; ‡, significantly (P < .05) different
from dogs receiving glyburide and isoflurane. (From Kersten JR, Schmeling
TJ, Hettrick DA, et al: Mechanism of cardioprotection by isoflurane: Role of adenosine
triphosphate-regulated potassium (KATP) channels. Anesthesiology 85:794–807,
1996.)
Figure 7-19
Percent segment shortening (%SS) in the intermittently
ischemic and reperfused left anterior descending coronary artery (LAD) region. The
%SS decreases significantly (P < .05) from baseline
during each 5-minute LAD occlusion and reperfusion in all groups. Significant decreases
in %SS during each 5-minute reperfusion and throughout 180 minutes of final reperfusion
are observed in dogs pretreated with glyburide, a KATP
channel antagonist,
in the presence or absence of isoflurane. The %SS recovers to baseline values after
reperfusion in dogs receiving isoflurane alone. †, Significantly (P
< .05) different from dogs pretreated with drug vehicle; §, significantly
(P < .05) different from dogs pretreated with
glyburide; ‡, significantly (P < .05) different
from dogs receiving glyburide and isoflurane. (From Kersten JR, Schmeling
TJ, Hettrick DA, et al: Mechanism of cardioprotection by isoflurane: Role of adenosine
triphosphate-regulated potassium (KATP) channels. Anesthesiology 85:794–807,
1996.)
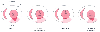 Figure 7-20
Schematic illustration of canine myocardium subjected
to a 60-minute coronary artery occlusion and reperfusion and then stained to identify
the region of myocardial infarction (black area)
within the area of myocardium at risk for infarction (light
gray area). Isoflurane decreased the extent of myocardial infarction.
The protective effect of isoflurane was equivalent to that produced by ischemic
preconditioning and was abolished by pretreatment with glyburide. *, Significantly
(P < .05) different from the control. (Adapted
from Kersten JR, Schmeling TJ, Pagel PS, et al: Isoflurane mimics ischemic preconditioning
via activation of KATP
channels. Reduction of myocardial infarct size
with an acute memory phase. Anesthesiology 87:361–370, 1997.)
Figure 7-20
Schematic illustration of canine myocardium subjected
to a 60-minute coronary artery occlusion and reperfusion and then stained to identify
the region of myocardial infarction (black area)
within the area of myocardium at risk for infarction (light
gray area). Isoflurane decreased the extent of myocardial infarction.
The protective effect of isoflurane was equivalent to that produced by ischemic
preconditioning and was abolished by pretreatment with glyburide. *, Significantly
(P < .05) different from the control. (Adapted
from Kersten JR, Schmeling TJ, Pagel PS, et al: Isoflurane mimics ischemic preconditioning
via activation of KATP
channels. Reduction of myocardial infarct size
with an acute memory phase. Anesthesiology 87:361–370, 1997.)
Volatile anesthetics may activate parallel or redundant signal transduction pathways that involve KATP channel opening to generate a physiologically meaningful cellular response. The sequential activation of several intracellular elements within a given transduction pathway may facilitate signal amplification and interaction between other redundant signaling systems. For example, administration of isoflurane in the presence of the KATP channel opener nicorandil[333] or diazoxide[343] markedly enhanced protection against ischemic injury beyond that observed with either drug alone. The combination of isoflurane and a selective δ1 -opioid receptor agonist amplified the preconditioning response in the rat.[343] This effect was synergistic and was sensitive to inhibition by glyburide.[343] The combined administration of isoflurane and morphine also markedly reduced infarct size in vivo,[332] and this protective effect was abolished by 5-HD.[332] These data suggest that combined administration of a volatile anesthetic and an opioid capable of agonist action at the δ1 -receptor subtype may stimulate similar or cooperative signaling cascades that amplify KATP channel activation to profoundly augment myocardial protection beyond that produced by either drug alone.
KATP channels in vascular smooth muscle cells are essential regulators of coronary vascular tone when ATP production is reduced.[344] Volatile anesthetic-induced coronary vasodilation[278] [345] [346] [347] [348] is attenuated by glyburide, indicating an important role for KATP channels in this process. It is possible that the beneficial actions of APC may be partially attributed to increases in myocardial oxygen supply mediated by KATP channel-dependent coronary vasodilation. However, sevoflurane increases coronary collateral blood flow in the presence of glyburide in vivo, indicating that this volatile anesthetic may enhance coronary collateral blood flow independent of KATP channel activation.[349] Findings demonstrate that sevoflurane-induced increases in collateral perfusion occur as a result of activation of Ca2+ -regulated potassium (BKCa ) and not KATP channels.[350] Isoflurane and sevoflurane directly activate mitochondrial KATP channels in isolated cardiac myocytes, and this action is linked to enhanced cell viability.[329] Based on these and other data collected from in vitro studies, it appears highly unlikely that myocardial protection produced by volatile anesthetics is solely related to favorable alterations in coronary vascular tone mediated by KATP channels.
Several receptor-mediated events and intracellular signaling elements that converge on the KATP channel have been implicated in APC. Pertussis toxin abolished reductions in infarct size produced by isoflurane ( Fig. 7-21 ), indicating that inhibitory guanine (Gi ) nucleotide-binding proteins are linked to the signal transduction pathways that mediate APC.[351] In contrast, pertussis toxin did not prevent the beneficial effects of direct KATP channel opening produced by nicorandil. These data strongly support the contention that volatile anesthetics modulate KATP channel activity through a second messenger. Halothane-induced
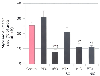 Figure 7-21
Histograms depict myocardial infarct size as a percentage
of area at risk in dogs pretreated with vehicle (Control) or pertussis toxin (PTX)
in the presence or absence of 1.0 minimum alveolar concentration of isoflurane (ISO)
or nicorandil (NIC). *, Significantly (P <
.05) different from the control; †, significantly (P
< .05) different from PTX alone; ‡, significantly (P
< .05) different from PTX plus ISO. (Adapted from Toller WG, Kersten
JR, Gross ER, et al: Isoflurane preconditions myocardium against infarction via
activation of inhibitory guanine (Gi) nucleotide binding proteins. Anesthesiology
92:1400–1407, 2000.)
Figure 7-21
Histograms depict myocardial infarct size as a percentage
of area at risk in dogs pretreated with vehicle (Control) or pertussis toxin (PTX)
in the presence or absence of 1.0 minimum alveolar concentration of isoflurane (ISO)
or nicorandil (NIC). *, Significantly (P <
.05) different from the control; †, significantly (P
< .05) different from PTX alone; ‡, significantly (P
< .05) different from PTX plus ISO. (Adapted from Toller WG, Kersten
JR, Gross ER, et al: Isoflurane preconditions myocardium against infarction via
activation of inhibitory guanine (Gi) nucleotide binding proteins. Anesthesiology
92:1400–1407, 2000.)
IPC produces translocation and phosphorylation of several protein kinases, most importantly PKC, involved in signal transduction.[356] [357] [358] PKC is an essential component of the signaling pathway involved in protecting
Reperfusion of ischemic myocardium is associated with the release of large quantities of reactive oxygen species (ROS)[369] [370] [371] [372] that disrupt intracellular Ca2+ homeostasis, cause lipid peroxidation, damage cell membranes, depress contractility, and produce reversible and irreversible tissue injury. Halothane, isoflurane, and enflurane can attenuate the toxic effects of oxygen-derived free radicals on LV pressure development in isolated hearts.[308] Isoflurane decreased hydroxyl radical generation in the ischemic rat heart,[373] and halothane had a similar effect in dogs.[374] The protective effects of sevoflurane were associated with reduced dityrosine formation, an indirect marker of reactive oxygen and nitrogen species.[375] These results support the contention that volatile anesthetics may reduce the release of deleterious quantities of ROS immediately after coronary artery occlusion and reperfusion.
In contrast to the data implicating a pathologic role of large amounts of ROS, newer evidence strongly suggests that a variety of preconditioning stimuli, including brief ischemia, mitochondrial KATP channel openers, opioids, and volatile anesthetics, stimulate a small burst of ROS that paradoxically appears to initiate downstream signaling events and produce protection from subsequent ischemic injury. The beneficial actions of sevoflurane against ischemic damage were abolished by scavengers of superoxide anion and inhibition of nitric oxide synthase. [375] These results suggest that superoxide anion may act to trigger APC and indicate that nitric oxide may scavenge superoxide anion on reperfusion to reduce injury. ROS scavengers attenuated isoflurane-induced reductions in myocardial infarct size in rabbits.[376] [377] Similarly, ROS inhibited the salutary effects of IPC[378] [379] or mitochondrial KATP channel openers. [380] Isoflurane directly increases superoxide formation in vivo independent of ischemia and reperfusion.[377] Volatile anesthetics may be capable of producing small amounts of ROS that exert protective effects during subsequent ischemia. These data provide compelling evidence that small quantities of ROS play a critical role in APC.
Activation of mitochondrial KATP channels is associated with the ROS generation.[381] Mitochondrial KATP channel opening produced by selective agonists generated ROS that appeared to be essential for activation of MAP kinase[382] and the subsequent beneficial effects against ischemic injury.[383] Morphine increased the fluorescence intensity of the hydrogen peroxide-sensitive probe 2',7'-dichloro-fluorescin,[384] actions that were blocked by 5-HD. These data also demonstrate a link between activation of mitochondrial KATP channels by opioids and ROS production. Mitochondria have been hypothesized to be a source of ROS production,[379] [385] [386] [387] [388] but whether opening of mitochondrial KATP channels directly contributes to free radical generation remains unclear. Conversely, ROS may also modulate KATP channel activity.[389] Isoflurane-induced increases in ROS are abolished by the mitochondrial KATP channel antagonist 5-HD, [390] further suggesting a link between ROS generation and mitochondrial KATP channel activation during APC.
The identities of the ROS involved in IPC and APC and the signaling pathways that are modulated by these free radicals also remain unclear. ROS have been shown to activate PKC, restore contractility, and limit the extent of myocardial infarction in rabbit hearts.[391] Hydrogen peroxide stimulates tyrosine kinase-dependent activation of phospholipase C in mouse embryonic fibroblasts, rendering these cells resistant to stress.[392] ROS may also directly stimulate PKC activity[393] or indirectly enhance the activity of the enzyme by activation of PLC.[392] Hydrogen peroxide activates Gi and Go proteins,[394] [395] as well as other protein kinases involved in reducing cellular injury.[396] [397] Hydrogen peroxide may also be converted to more reactive species that subsequently modify cysteine residues specific to Gαi and Gαo and selectively activate these proteins.[395] A strong possibility exists that volatile anesthetic-induced production of ROS may directly activate several intracellular mediators of endogenous protection against ischemic injury. However, this tantalizing hypothesis requires further investigation.
|
|
|
|
|
|
|
|
|
|
|
|
|