|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The brain has a high rate of energy utilization and very limited energy storage capacity. It is therefore extremely
 Figure 21-13
Relationships between cerebral perfusion, cerebral blood
flow (CBF), the electroencephalogram (EEG), and the functional status/viability of
neurons. Note that in the approximate CBF range of 6 to 12 mL/kg/min, the energy
supply is insufficient to support electrophysiologic activity (i.e., EEG flat), but
it can prevent complete membrane failure and neuronal death for extended periods.
These areas are referred to as the ischemic penumbra.[437]
The data are derived from studies in the cerebral cortex of barbiturate-anesthetized
baboons[437]
[438]
and unanesthetized monkeys.[439]
The CBF and mean
arterial pressure thresholds may vary with the anesthetic and species.[440]
Figure 21-13
Relationships between cerebral perfusion, cerebral blood
flow (CBF), the electroencephalogram (EEG), and the functional status/viability of
neurons. Note that in the approximate CBF range of 6 to 12 mL/kg/min, the energy
supply is insufficient to support electrophysiologic activity (i.e., EEG flat), but
it can prevent complete membrane failure and neuronal death for extended periods.
These areas are referred to as the ischemic penumbra.[437]
The data are derived from studies in the cerebral cortex of barbiturate-anesthetized
baboons[437]
[438]
and unanesthetized monkeys.[439]
The CBF and mean
arterial pressure thresholds may vary with the anesthetic and species.[440]
Much has been made of the difference between complete cerebral ischemia, as occurs during cardiac arrest, and incomplete cerebral ischemia, as may occur during occlusion of a major cerebral vessel or severe hypotension. However, from the clinician's vantage, the important difference is that the residual blood flow during incomplete ischemia may result in enough oxygen delivery to allow for some generation of ATP and thereby stave off the catastrophic irreversible membrane failure that occurs within minutes during normothermic complete cerebral ischemia. This difference in the rate of failure of the energy supply[446] [447] ( Fig. 21-14 ) can result in a much greater apparent tolerance for focal or incomplete ischemia than for complete global ischemia (e.g., cardiac arrest). The difference is in reality a matter of severity rather than the actual initial pathophysiology of the insult, with the exception that limited residual perfusion may potentially be detrimental in the event of hyperglycemia. There is also a theoretical concern that low levels of residual flow will provide sufficient oxygen delivery to permit the formation of oxygen radicals. However, the observation that focal ischemia is better tolerated than complete global ischemia argues against major importance.
Energy failure is the central event that occurs during cerebral ischemia.[448] ATP is required for maintenance of the normal membrane ionic gradient, and energy failure rapidly ensues with membrane depolarization and influx of sodium and calcium into the neuron. Voltage-dependent
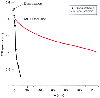 Figure 21-14
Comparison of the rates of failure of the energy supply
(adenosine triphosphate [ATP]) in complete global ischemia (produced by decapitation
in dogs[447]
) and incomplete focal ischemia (middle
cerebral artery occlusion [MCA] in monkeys[446]
).
In the presence of residual CBF, failure of the energy supply is substantially delayed.
Figure 21-14
Comparison of the rates of failure of the energy supply
(adenosine triphosphate [ATP]) in complete global ischemia (produced by decapitation
in dogs[447]
) and incomplete focal ischemia (middle
cerebral artery occlusion [MCA] in monkeys[446]
).
In the presence of residual CBF, failure of the energy supply is substantially delayed.
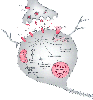 Figure 21-15
During ischemia, depletion of adenosine triphosphate
leads to neuronal depolarization and the subsequent release of supranormal quantities
of neurotransmitters, especially glutamate. Excessive stimulation of ligand-gated
channels and the simultaneous opening of voltage-dependent Ca2+
channels
permits rapid entry of Ca2+
into neurons. Stimulation of metabotropic
glutamate receptors (mGlu) generates inositol triphosphate (IP3
), which
causes release of Ca2+
from the endoplasmic reticulum (ER)/mitochondria.
Activation of the α-amino-3-hydroxy-5-methyl-4-isoxazopropionic acid (AMPA)-gated
subset of glutamate receptors also permits excessive entry of sodium (Na+
).
Excessive free Ca2+
results in the activation of numerous enzymes: protease
activation causes breakdown of the cytoskeleton of the neuron; lipases damage plasma
membrane lipids and release arachidonic acid, which is metabolized by cyclooxy-genases
and lipoxygenases to yield free radicals and other mediators of cell injury; activation
of nitric oxide synthase (NOS) leads to release of nitric oxide (NO) and, in turn,
the generation of peroxynitrite, a highly reactive free radical; and activated endonucleases
damage DNA, thereby rendering the neuron susceptible to apoptosis. Injury to mitochondria
leads to energy failure, free radical generation, and the release of cytochrome c
(Cyt c) from mitochondria; the latter is one of the means by which neuronal apoptosis
is initiated. NMDA, N-methyl-D-aspartate;
ROS, reactive oxygen species; VGCC, voltage-gated calcium channels.
Figure 21-15
During ischemia, depletion of adenosine triphosphate
leads to neuronal depolarization and the subsequent release of supranormal quantities
of neurotransmitters, especially glutamate. Excessive stimulation of ligand-gated
channels and the simultaneous opening of voltage-dependent Ca2+
channels
permits rapid entry of Ca2+
into neurons. Stimulation of metabotropic
glutamate receptors (mGlu) generates inositol triphosphate (IP3
), which
causes release of Ca2+
from the endoplasmic reticulum (ER)/mitochondria.
Activation of the α-amino-3-hydroxy-5-methyl-4-isoxazopropionic acid (AMPA)-gated
subset of glutamate receptors also permits excessive entry of sodium (Na+
).
Excessive free Ca2+
results in the activation of numerous enzymes: protease
activation causes breakdown of the cytoskeleton of the neuron; lipases damage plasma
membrane lipids and release arachidonic acid, which is metabolized by cyclooxy-genases
and lipoxygenases to yield free radicals and other mediators of cell injury; activation
of nitric oxide synthase (NOS) leads to release of nitric oxide (NO) and, in turn,
the generation of peroxynitrite, a highly reactive free radical; and activated endonucleases
damage DNA, thereby rendering the neuron susceptible to apoptosis. Injury to mitochondria
leads to energy failure, free radical generation, and the release of cytochrome c
(Cyt c) from mitochondria; the latter is one of the means by which neuronal apoptosis
is initiated. NMDA, N-methyl-D-aspartate;
ROS, reactive oxygen species; VGCC, voltage-gated calcium channels.
Calcium is a ubiquitous second messenger in cells and is a cofactor required for the activation of a number of enzyme systems. The rapid, uncontrolled increase in cytosolic Ca levels initiates the activation of a number of cellular processes that contribute to injury. Cytoskeletal proteins such as actin are cleaved by activated proteases.[450] These enzymes also degrade a number of the protein
DNA damage is also an important event during ischemic neuronal injury. Generation of free radicals from arachidonic acid metabolism, from injured mitochondria, and from the production of peroxynitrite from NO leads to oxidative injury to DNA.[452] Activation of endonucleases also produces DNA strand breaks. Under normal circumstances, DNA injury results in the activation of poly-ADP ribose-polymerase (PARP), an enzyme that participates in DNA repair. With excessive DNA injury, PARP activity increases dramatically, and this increased activity can lead to depletion of nicotinamide adenine dinucleotide (NAD), a substrate of PARP.[452] NAD is also an important coenzyme in energy metabolism, and its depletion further exacerbates energy failure.
Lactate formation is an additional element of the pathophysiologic process. Lactic acid is formed as a result of the anaerobic glycolysis that takes place after failure of the supply of oxygen. The associated decline in pH contributes to deterioration of the intracellular environment. An increased preischemic serum glucose level may accelerate this process by providing additional substrate for anaerobic glycolysis.[453] [454] [455] [456]
NO,[2] [3] [457] which has emerged as a probable mediator of CBF changes in many normal physiologic states (see the preceding sections), is also of relevance to the pathophysiology of ischemia. NO is, in fact, a weak free radical [457] [458] that in turn leads to the generation of a more reactive species (peroxynitrite), and it is a "killer substance" used by macrophages.[2] In cerebral ischemia, NO is probably both friend and foe.[459] It is likely that during a period of focal ischemia, the vasodilating effect of NO (probably constitutively elaborated NO of endothelial origin) serves to augment collateral CBF.[460] However, in the postischemic phase, NO (probably inducible NO of neuronal origin) contributes to neuronal injury. [460]
Collectively, the simultaneous and unregulated activation of a number of cellular pathways overwhelms the reparative and restorative processes within the neuron and ultimately leads to neuronal death.
The neuronal death that occurs in response to these processes has been categorized as necrotic or apoptotic in nature. Necrotic death of neurons, mediated by excitotoxic injury, is characterized by rapid cellular swelling, condensation and pyknosis of the nucleus, and swelling of the mitochondria and endoplasmic reticulum. A characteristic of these necrotic neurons is the presence of an acidophilic cytoplasm. [461] Necrotic neuronal death results in local infiltration of the brain by inflammatory cells. A consequence of this inflammation is a considerable amount of collateral damage.
Neuronal apoptosis, a form of cellular suicide, has also been demonstrated in a variety of models of cerebral ischemia. Apoptosis is characterized by condensation of chromatin, involution of the cell membrane, swelling of mitochondria, and cellular shrinkage. In the later stages of apoptosis, neurons fragment into several apoptotic bodies, which are then cleared from the brain.[461] The lack of a substantial inflammatory response to apoptotic death limits injury to the surrounding neurons that have survived the initial ischemic insult.
A number of biochemical pathways that lead to apoptosis have been described. Initiation of apoptosis by the release of cytochrome c from injured mitochondria has been studied the most ( Fig. 21-16 ). Cytochrome c is restricted from the cytoplasm by the outer mitochondrial membrane.[462] When mitochondria are injured, pores within the outer membrane allow cytochrome c to be released into the cytoplasm, where it interacts with procaspase-9 and apoptosis-activating factor to produce an apoptosome.[463] Procaspase-9 undergoes activation by proteolytic cleavage. Activated caspase-9 then activates caspase-3. The latter serves as an executor of apoptosis by cleaving a number of protein substrates that are essential in DNA repair (such as PARP). Activation of caspase-3 can also occur by inflammatory signaling through activation of tumor necrosis factor-α (TNF-α) and caspase-8.[464] It should be noted that the neuronal injury that occurs in response to ischemia cannot easily be divided into necrosis or apoptosis. The nature of neuronal death probably encompasses a spectrum in which some neurons undergo necrosis or apoptosis whereas others undergo cell death that has features of both necrosis and apoptosis.
The traditional concept of ischemic injury was that neuronal death was restricted to the time of ischemia and during the early reperfusion period. However, more recent data indicate that postischemic neuronal injury is a dynamic process in which neurons continue to die for a long period after the initiating ischemic insult ( Fig. 21-17 ).[465] [466] [467] This delayed neuronal death, which was first demonstrated in models of global cerebral ischemia, has been demonstrated during focal ischemia as well. The extent of delayed neuronal death depends on the severity of the ischemic insult. With severe ischemia, most neurons undergo rapid death. With more moderate insults, neurons that survive the initial insult undergo delayed death. This ongoing neuronal loss contributes to the gradual expansion of cerebral infarction after focal ischemia. In experimental studies, evidence of cerebral inflammation, which can theoretically contribute to further injury, has been demonstrated even 6 to 8 months after the primary ischemia.
The occurrence of delayed neuronal death has important implications for the evaluation of studies in which neuroprotective strategies are investigated. A wide variety of interventions have shown neuroprotective efficacy in studies in which the extent of injury is evaluated within 3 to 4 days after ischemia. However, this neuroprotective efficacy may not be sustained. Recent data indicate that
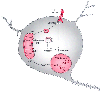 Figure 21-16
Cellular processes that lead to neuronal apoptosis.
Cytochrome c (cyt c), which is normally restricted
to the space between the inner and outer mitochondrial membranes, is released in
response to mitochondrial injury. Cytochrome c,
in combination with apoptosis activating factor (apaf), activates caspase-9 by proteolytic
cleavage. Activated caspase-9 then leads to activation of caspase-3. This enzyme
cleaves a number of substrates, including those necessary for DNA repair. Within
the mitochondria, bax augments and bcl prevents the release of cytochrome c.
Cytochrome c release can also be initiated by bid,
a substance that is activated by caspase-8 through tumor necrosis factor-α
(TNF) signaling. In addition, caspase-8 can directly activate caspase 3. Excessive
activation of poly-ADP ribose-polymerase (PARP), an enzyme integral to DNA repair,
depletes cellular stores of oxidized nicotinamide adenine dinucleotide (NAD+
).
Depletion of NAD+
further exacerbates the energy failure because it is
central to energy metabolism. ATP, adenosine triphosphate.
Figure 21-16
Cellular processes that lead to neuronal apoptosis.
Cytochrome c (cyt c), which is normally restricted
to the space between the inner and outer mitochondrial membranes, is released in
response to mitochondrial injury. Cytochrome c,
in combination with apoptosis activating factor (apaf), activates caspase-9 by proteolytic
cleavage. Activated caspase-9 then leads to activation of caspase-3. This enzyme
cleaves a number of substrates, including those necessary for DNA repair. Within
the mitochondria, bax augments and bcl prevents the release of cytochrome c.
Cytochrome c release can also be initiated by bid,
a substance that is activated by caspase-8 through tumor necrosis factor-α
(TNF) signaling. In addition, caspase-8 can directly activate caspase 3. Excessive
activation of poly-ADP ribose-polymerase (PARP), an enzyme integral to DNA repair,
depletes cellular stores of oxidized nicotinamide adenine dinucleotide (NAD+
).
Depletion of NAD+
further exacerbates the energy failure because it is
central to energy metabolism. ATP, adenosine triphosphate.
 Figure 21-17
Time course of neuronal death. Excitotoxic (glutamate-mediated)
injury results in neuronal death within the first few hours after the onset of ischemia.
Brain tissue injury elicits an inflammatory response, an important process in removal
of injured tissue and healing, that leads to a substantial amount of collateral damage.
Inflammation-mediated neuronal death can continue for several days. Neuronal apoptosis
can occur in injured neurons that survived the initial insult. Apoptotic neuronal
death has been demonstrated to occur for many days after the initiating ischemic
insult. It is now apparent that ischemic neuronal death is a dynamic process in
which neurons continue to die for a long period. (Adapted from Dirnagl U,
Iadecola, C, Moskowitz MA: Pathobiology of ischaemic stroke: An integrated view.
Trends Neurosci 22:391–397, 1999.)
Figure 21-17
Time course of neuronal death. Excitotoxic (glutamate-mediated)
injury results in neuronal death within the first few hours after the onset of ischemia.
Brain tissue injury elicits an inflammatory response, an important process in removal
of injured tissue and healing, that leads to a substantial amount of collateral damage.
Inflammation-mediated neuronal death can continue for several days. Neuronal apoptosis
can occur in injured neurons that survived the initial insult. Apoptotic neuronal
death has been demonstrated to occur for many days after the initiating ischemic
insult. It is now apparent that ischemic neuronal death is a dynamic process in
which neurons continue to die for a long period. (Adapted from Dirnagl U,
Iadecola, C, Moskowitz MA: Pathobiology of ischaemic stroke: An integrated view.
Trends Neurosci 22:391–397, 1999.)
|
|
|
|
|
|
|
|
|
|
|
|
|