 |
|
|
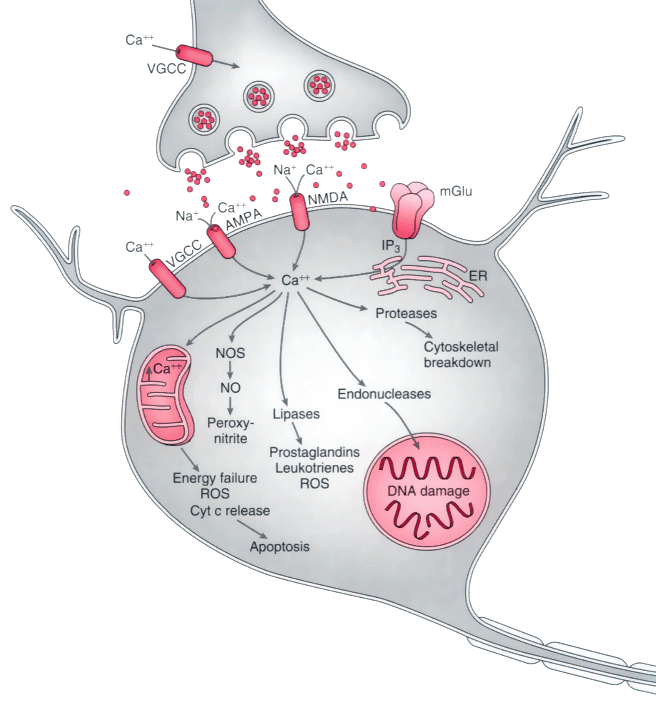
Figure 21-15
During ischemia, depletion of adenosine triphosphate
leads to neuronal depolarization and the subsequent release of supranormal quantities
of neurotransmitters, especially glutamate. Excessive stimulation of ligand-gated
channels and the simultaneous opening of voltage-dependent Ca2+
channels
permits rapid entry of Ca2+
into neurons. Stimulation of metabotropic
glutamate receptors (mGlu) generates inositol triphosphate (IP3
), which
causes release of Ca2+
from the endoplasmic reticulum (ER)/mitochondria.
Activation of the α-amino-3-hydroxy-5-methyl-4-isoxazopropionic acid (AMPA)-gated
subset of glutamate receptors also permits excessive entry of sodium (Na+
).
Excessive free Ca2+
results in the activation of numerous enzymes: protease
activation causes breakdown of the cytoskeleton of the neuron; lipases damage plasma
membrane lipids and release arachidonic acid, which is metabolized by cyclooxy-genases
and lipoxygenases to yield free radicals and other mediators of cell injury; activation
of nitric oxide synthase (NOS) leads to release of nitric oxide (NO) and, in turn,
the generation of peroxynitrite, a highly reactive free radical; and activated endonucleases
damage DNA, thereby rendering the neuron susceptible to apoptosis. Injury to mitochondria
leads to energy failure, free radical generation, and the release of cytochrome c
(Cyt c) from mitochondria; the latter is one of the means by which neuronal apoptosis
is initiated. NMDA, N-methyl-D-aspartate;
ROS, reactive oxygen species; VGCC, voltage-gated calcium channels.
|
|