|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The structure and function of the sympathetic and parasympathetic systems are discussed in the preceding sections. Pharmacologic manipulation of autonomic function is the basis of therapy in many acute and
The endogenous sympathetic transmitters norepinephrine, epinephrine,
and dopamine are catecholamines, an important subclass of the sympathomimetic drugs
( Fig. 16-20
). The parent
compound of this sympathomimetic group is β-phenylethylamine, the structure
of which includes a benzene ring and ethylamine side chain. Substitution of hydroxyl
groups at the 3 and 4 positions of the benzene ring converts benzene to catechol,
and these compounds are known as catecholamines.
Although synthetic, isoproterenol and dobutamine are
| Site of Action | Compounds that Augment Autonomic Activity | Compounds that Depress Autonomic Activity * |
|---|---|---|
| Sympathetic and parasympathetic ganglia | Stimulate postganglionic neurons | Block conduction |
|
|
Nicotine | Hexamethonium (C-6) |
|
|
Inhibit AChE | Mecamylamine (Inversine) |
|
|
Physostigmine (Eserine) | Trimethaphan (Arfonad) |
|
|
Neostigmine (Prostigmin) | High concentrations of ACh |
|
|
Parathion | Anticholinesterase drugs |
| Endings of postganglionic noradrenergic neurons | Release NE | Block NE synthesis |
|
|
Tyramine | Metyrosine (Demser) |
|
|
Ephedrine | Interfere with NE storage |
|
|
Amphetamine | Reserpine |
|
|
|
Guanethidine (Ismelin) |
|
|
|
Prevent NE release |
|
|
|
Bretylium (Bretylol) |
|
|
|
Guanethidine (Ismelin) |
|
|
|
Form false transmitters |
|
|
|
Methyldopa (Aldomet) |
| α-Receptors | Stimulate α1 -receptors | Block α-receptors |
|
|
Methoxamine (Vasoxyl) | Phenoxybenzamine (Dibenzyline) |
|
|
Phenylephrine (Neo-Synephrine) |
|
|
|
Stimulate α2 -receptors | Phentolamine (Regitine) |
|
|
Clonidine † (Catapres) | Prazosin (Minipres) (blocks α1 ) |
|
|
|
Yohimbine (blocks α2 ) |
| β-Receptors | Stimulate β-receptors | Block β-receptors |
|
|
Isoproterenol (Isuprel) | Propranolol (Inderal) and others (block β1 and β2 ) |
|
|
Dobutamine (Dobutrex) |
|
|
|
|
Atenolol (Tenormin) and others (block β1 ) |
| ACh, acetylcholine; NE, norepinephrine; AChE, acetylcholinesterase. | ||
| Adapted from Ganong W: The autonomic nervous system. In Ganong W (ed): Review of Medical Physiology, 15th ed. Norwalk, CT, Appleton & Lange, 1991, p 210. | ||
The catecholamines are primarily metabolized by COMT. The loss of either hydroxyl group enhances oral effectiveness and duration of action because the drug is no longer metabolized by COMT. Noncatecholamines are primarily metabolized by MAO. The noncatecholamines that have a substituted α-carbon have a longer duration of action because they are not metabolized by COMT or MAO.[163]
Epinephrine is used intravenously in life-threatening circumstances, including the treatment of cardiac arrest, circulatory collapse, and anaphylaxis, but it is also commonly used locally to limit the spread of local anesthetics or to reduce blood loss. The systemic effects of epinephrine are variable and are related to blood levels. The choice of dosing and the route of administration are determined by the indication for use and its urgency.
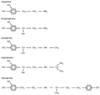 Figure 16-20
Catecholamine structures. A benzene ring with two adjacent
hydroxyl groups forms the catechol nucleus.
Figure 16-20
Catecholamine structures. A benzene ring with two adjacent
hydroxyl groups forms the catechol nucleus.
Epinephrine activates all adrenergic receptors: α1 , α2 , β1 , β2 , and β3 . Potential therapeutic effects of epinephrine include positive inotropy, chronotropy, and enhanced conduction in the heart (β1 ); smooth muscle relaxation in the vasculature and bronchial tree (β2 ); and vasoconstriction (α1 ). With vasoconstriction, aortic diastolic pressure is increased, promoting coronary flow during cardiac arrest, which may be the single most important determinant of survival.[164] Endocrine and metabolic effects of epinephrine include increased levels of glucose, lactate, and free fatty acids (see Table 16-1 ).
Epinephrine may be given intravenously as a bolus or by infusion. Usual bolus doses for pressure support begin at 2 to 8 µg given intravenously; 0.02 mg/kg or approximately 1.0 mg is given for cardiovascular collapse, asystole, ventricular fibrillation, electromechanical dissociation, or anaphylactic shock. The higher dose range is recommended in these critical situations to maintain myocardial and cerebral perfusion through peripheral vasoconstriction. High-dose epinephrine (0.1 to 0.2 mg/kg) has been studied in resuscitation from cardiac arrest, but it does not appear to improve rates of survival in adults. High-dose epinephrine may be given in adults if usual doses fail to generate a response.[165] In pediatric patients, outcome after asystole and pulseless cardiac arrest is abysmal, and the current recommendation is that high-dose epinephrine (0.1 mg/kg) be administered within 3 to 5 minutes after the initial dose (0.01 mg/kg) and repeated every 3 to 5 minutes throughout resuscitative attempts.[166] Doses as large as 0.2 mg/kg may be effective.[167] Endotracheal or intraosseous administration is an option in urgent situations such as cardiac arrest while intravenous access is obtained. Endotracheal doses should be at least tripled and diluted in 10 mL of normal saline in adult patients.[168] In pediatric patients, 10 times the intravenous dose may be needed.[166] When continued cardiovascular support is required, infusion rates can be adjusted to elicit specific receptor stimulation. Epinephrine should not be administered in alkaline solutions, because it is rapidly degraded to its biologically inactive metabolite, adrenochrome. If adrenochrome is present, the vial will have a pink hue and should be discarded.[163]
Patients vary tremendously in their response to these agents, and given rates of infusion cannot guarantee the expected serum levels in all patients; the "pressors" therefore should be carefully titrated, and measures to monitor renal, cerebral, and myocardial perfusion are
In the past, inhaled epinephrine was used in a 1% (1 g/100 mL) solution to treat bronchospasm, but it has been largely supplanted by β2 -specific agonists. Racemic epinephrine (i.e., mixture of the levorotary and dextrorotary isomers) constricts edematous mucosa and is used in the treatment of severe croup [169] and of postextubation and traumatic airway edema. A 2.25% solution (MicroNefrin or Vaponefrin) is diluted with water or saline in a 1:8 ratio and is nebulized. Treatments may be given as frequently as every 2 hours, with effects lasting 30 to 60 minutes; the patient should remain under observation for at least 2 hours, because initial improvement may be followed by rebound swelling up to 2 hours after administration.[163] Although it is common clinical practice to use the racemic form of epinephrine for these clinical applications, data show that L-epinephrine is 15 to 30 times more potent than the mixture[170] [171] and is equally effective and less expensive in treating these clinical complications. [172]
Bronchospasm may also be treated by subcutaneous administration
of epinephrine in doses of 300 µg every
| Drug * (Proprietary Name) | Receptors | Usual Infusion Rate |
|---|---|---|
| Epinephrine (Adrenalin) | β2 | 1–2 µg/min |
|
|
β1 + β2 | 2–10 µg/min |
|
|
α1 | ≥10 µg/min † (bolus: 2–10 µg; 0.5–1.0 mg ‡ ) |
| Norepinephrine (Levophed) | α1 , β1 , ≫ β2 | 4–12 µg/min † |
| Dopamine (Intropin) | Dopaminergic | 0–3 µg/kg/min |
|
|
β | 3–10 µg/kg/min |
|
|
α | >10 µg/kg/min † |
| Dobutamine (Dobutrex) | β1 ≫ β2 , α | 2.5–10 µg/kg/min † |
| Isoproterenol (Isuprel) | β1 > β2 | 0.5–10 µg/min |
| Amrinone (Inocor) | Increase cyclic adenosine monophosphate through phosphodiesterase inhibition | 0.75 mg/kg/load over 2–3 min |
|
|
|
5–10 µg/kg/min infusion |
| Data from Hoffman BB, Lefkowitz RJ: Catecholamines and sympathetic drugs. In Goodman A, Rall T, Nies A, et al (eds): Goodman and Gilman's the Pharmacological Basis of Therapeutics, 8th ed. New York, Pergamon Press, 1990, p. 187. | ||
Epinephrine is often applied locally to mucosal surfaces to decrease bleeding in the operative site. It is mixed with local anesthetics for infiltration into tissues or intrathecal injection. The α-mediated vasoconstriction decreases bleeding in the area and slows vascular uptake of local anesthetic, prolonging the duration of effect and decreasing the peak serum level of the local anesthetic. Although clinicians express concern about the systemic effects of such injections, several studies have shown that, barring intravascular injection, the elevations in plasma levels from vascular uptake are relatively modest and are substantially less than levels seen during psychological stress.[29] [174]
Drug interactions with epinephrine are often predictable. Cocaine and other uptake inhibitors enhance the effect and duration of exogenous epinephrine. Preexisting α1 -blockade can cause the paradoxic phenomenon of epinephrine reversal as the β2 -vasodilating effects are unmasked. Patients receiving nonselective β-blockers may demonstrate unopposed α-responses. Cardioselective (β1 ) blockade does not have this effect.[175]
Halothane sensitizes the heart to catecholamines, and the potential for troublesome arrhythmias under light inhalational anesthesia has long been appreciated by clinicians. Epinephrine decreases the refractory period, rendering the heart more susceptible to arrhythmias. In adults, the epinephrine dose required to produce three premature ventricular contractions in 50% of patients (ED50 ) at 1.25 minimum alveolar concentration (MAC) was 2.1 µg/kg for halothane, 6.7 µg/kg for isoflurane, and 10.9 µg/kg for enflurane.[176] Children appear to tolerate higher doses than do adults. It has been suggested that children receiving a halothane anesthetic can receive a maximum of 10 to 15 µg/kg of epinephrine subcutaneously every 10 minutes.[169] Hypocapnia potentiates this drug interaction.
Norepinephrine differs structurally from epinephrine only in its lack of a methyl group. Like epinephrine, norepinephrine acts at α- and β-receptors, but it is typically used for its potent α-agonism. It is frequently the pressor of last resort in supporting the systemic vascular resistance. Because of its short half-life of 2.5 minutes, a continuous infusion is preferred. Whereas less than 2 µg/min (30 ng/kg/min) may uncover the effects of β1 -adrenergic stimulation, the usual infusion rates of greater than 3 µg/min (50 ng/kg/min) elicit peripheral vasoconstriction from α-adrenergic stimulation.[177]
Peripheral vasoconstriction increases blood pressure and may cause reflex bradycardia. Venous return is increased by the powerful venoconstriction. Cardiac output is frequently unchanged or decreased; oxygen consumption is markedly increased. Pulmonary vascular resistance may be increased, and norepinephrine should be used with caution in patients with pulmonary hypertension.[178]
Like epinephrine, norepinephrine is a potent constrictor of the renal and mesenteric vascular beds and can cause renal failure, mesenteric infarction, and peripheral hypoperfusion. The decrease in hepatic flow is of clinical relevance because plasma levels of hepatically metabolized drugs (e.g., lidocaine) are markedly increased.[179] To ameliorate the renal effects, a low-dose dopamine infusion may be added to norepinephrine.[180] Extravasation of norepinephrine can cause tissue necrosis and may be treated with local infiltration of phentolamine. Prolonged infusion has caused gangrene of the digits. The potential for profound vasoconstriction makes careful patient selection and close monitoring mandatory.
Dopamine acts at α-adrenergic, β-adrenergic, and dopaminergic receptors; it also acts to release norepinephrine and therefore has mixed direct and indirect effects. Although dopamine is a precursor of norepinephrine, its most important effect may be to cause peripheral vasodilation. Improvement of blood flow through the renal and mesenteric beds in shocklike states is expected through its action at dopamine receptors on the postjunctional membrane. It is rapidly metabolized by MAO and COMT and has a half-life of about 1 minute. Like other endogenous catecholamines, it is given as a continuous intravenous infusion and without a loading dose. At low doses (0.5 to 2.0 µg/kg/min), DA1 receptors are stimulated, and renal and mesenteric vascular beds dilate.[181] In addition to an improvement in renal blood flow, glomerular filtration rate and sodium excretion increase. With an infusion rate of 2 to 10 µg/kg/min, β1 -receptors are stimulated, which increases cardiac contractility and output. Rates higher than 5 µg/kg/min stimulate release of endogenous norepinephrine, which contributes to cardiac stimulation. In larger doses (10 to 20 µg/kg/min), α- and β1 -receptors are stimulated, the α-adrenergic vasoconstrictive effect predominates, and the benefit to renal perfusion may be lost.[182] Patients' responses to dopamine are extremely variable, and dosages must be individualized. Monitoring to ensure organ and peripheral perfusion is critical. The dose should be significantly decreased in patients treated previously with an MAOI or tricyclic antidepressant.
Dopamine is frequently the initial agent used in the treatment of shock, particularly in vasodilated states such as sepsis; it is often used to protect the kidney and to aid in diuresis, especially in patients with severe CHF. [183] Infusion in combination with dobutamine for the therapy of cardiogenic shock may be more effective than either agent given alone. [184]
Dopexamine hydrochloride (Dopacard), an inotropic vasodilator, is a synthetic parenteral dopamine analog that may be of use in CHF. Dopexamine is approximately 60 times more potent at β2 -adrenergic receptors than dopamine, one third as potent at DA1 receptors, and one seventh as potent at DA2 receptors.[185] [186] Unlike dopamine, it shows no α-adrenergic effects and negligible β1 -adrenergic effects and is therefore devoid of vasoconstrictive activity.[185] [187] Dopexamine has a reported half-life of 3 to 7 minutes in healthy patients and approximately 11 minutes in patients with low cardiac output.[188] β2 -Agonism produces systemic vasodilation and indirect inotropic activity (through inhibition of neuronal uptake of norepinephrine).[185] [186] [187] [189] [190] The stimulation of dopaminergic receptors produces selective vasodilation of renal and splanchnic vessels and increases glomerular filtration rate, diuresis, and natriuresis. [186] [191] [192] [193] [194]
The use of dopexamine is preferable when vascular resistance is high. Within the dose range of 1 to 6 µg/kg/min, the combined inotropic, vasodilative, diuretic, and natriuretic effects have shown benefit in the management of CHF[195] [196] [197] [198] but an indeterminate outcome in the treatment of septic shock.[197] [199] [200] [201] [202] [203] Use of this agent has been limited by dose-dependent tachycardia, mainly at doses higher than 4 µg/kg/min.[204] [205] The effects of dopexamine on intestinal mucosal and hepatic perfusion remain controversial. [206] [207] [208] [209] [210] [211] In general, systemic vasodilation appears more pronounced with dopexamine, and positive inotropic effects appear more marked with dopamine[212] and dobutamine.[213]
Fenoldopam is a selective DA1 agonist and potent vasodilator (i.e., six to nine times as potent as dopamine) that enhances natriuresis, diuresis, and renal blood flow.[214] [215] [216] [217] [218] Because of its poor bioavailability and varied results in clinical trials, fenoldopam is no longer being investigated as a candidate for therapy of chronic hypertension or CHF. Instead, intravenous fenoldopam, given
Whereas the β-agonist isoproterenol and the α-agonists phenylephrine and methoxamine act predominantly at only one type of receptor, most of the sympathomimetic drugs act at α- and β-receptors. Most noncatecholamine sympathomimetic amines act at α- and β-receptors because they have two mechanisms of action: directly at a receptor and indirectly by releasing endogenous norepinephrine.
Mephentermine (Wyamine), ephedrine, and metaraminol (Aramine) are mixed-acting drugs. Ephedrine increases blood pressure and has a positive inotropic effect. Because it does not have detrimental effects on uterine blood flow, ephedrine is widely used as a pressor in the hypotensive parturient patient.[221] Because of its β1 -adrenergic stimulating effects, ephedrine is helpful in treating moderate hypotension, particularly if accompanied by bradycardia. It also has some direct β2 -adrenergic stimulating effects and has been used orally as a bronchodilator. The usual dose is 2.5 to 25 mg given intravenously or 25 to 50 mg administered intramuscularly. Mephentermine is similar to ephedrine in its effects, whereas metaraminol has relatively stronger direct α1 -adrenergic stimulating effects and may be associated with reflex bradycardia.
Tachyphylaxis to the indirect effect may develop through depletion
of norepinephrine stores. Although all sympathomimetic amines are capable of producing
tolerance or tachyphylaxis, the mechanism has been best studied with metaraminol.
Metaraminol is taken up into the sympathetic nerve ending, displacing norepinephrine
and producing the sympathomimetic effect. However, after some time, the drug acts
as a false transmitter, and subsequent sympathetic nerve stimulation results in much
less effect. Consequently, the drug probably should not be used widely when other,
more effective drugs are available. Indirect action is attenuated in the presence
of long-term reserpine or cocaine use, but these drugs may still be efficacious at
| Drug | Route | Bolus | Continuous Infusion | References |
|---|---|---|---|---|
| Clonidine | Oral | 150 µg, 4–5 µg/kg |
|
[230] [231] [234] [291] [304] |
|
|
Intramuscular | 2 µg/kg |
|
[272] |
|
|
Intravenous | 150 µg, 4–8 µg/kg | 2 µg/kg/hr | [266] [273] [305] |
|
|
Epidural | 150–450 µg, 6–8 µg/kg | 12.5–70 µg/hr, 1–2 µg/kg/hr | [240] [241] [242] [243] [270] [272] [282] [283] [284] [285] [286] [306] [307] [308] [309] |
|
|
Intrathecal | 30–225 µg | 8–400 µg/day | [244] [245] [246] [247] [248] [249] [301] [302] [303] [310] [311] [312] [313] [314] [315] [316] [317] |
| Dexmedetomidine | Intravenous | 1 µg/kg over 10 min | 0.4–0.7 µg/kg/hr | [261] |
Phenylephrine and methoxamine are selective α1 -agonists. These drugs are commonly used when peripheral vasoconstriction is needed and cardiac output is adequate, as in the hypotension that may accompany spinal anesthesia, and in patients with coronary artery disease or aortic stenosis to increase coronary perfusion pressure without chronotropic side effects. Phenylephrine (Neo-Synephrine) has a rapid onset and a relatively short duration of action (5 to 10 minutes) when given intravenously. It may be given by bolus doses of 40 to 100 µg or by infusion at a starting rate of 10 to 20 µg/min. Higher doses of up to 1 mg are used to slow supraventricular tachycardia through reflex action. Phenylephrine is also used as a mydriatic and nasal decongestant. In anesthetic practice, it is applied topically, alone or mixed with local anesthetic gel, to prepare the nose for intubation. It is also added to local anesthetic to prolong subarachnoid block. In contrast, methoxamine (Vasoxyl) is a much longer-acting drug (30 to 60 minutes). [223] In larger doses, methoxamine possesses some membrane-stabilizing and β-blocking properties.
The α2 -agonists are assuming greater importance as anesthetic adjuvants and analgesics. Their primary effect is sympatholytic. They reduce peripheral norepinephrine release by stimulation of prejunctional inhibitory α2 -adrenoreceptors. They inhibit central neural transmission in the dorsal horn by presynaptic and postsynaptic mechanisms and directly in spinal preganglionic sympathetic neurons. Traditionally, they have been used as antihypertensive drugs, but uses based on sedative, anxiolytic, and analgesic properties are being developed.
Clonidine, the prototypic drug of this class ( Table 16-10 ), is a selective partial agonist for α2 -adrenoreceptors, with a ratio of approximately 200:1 (α2 to α1 ). Its antihypertensive effects are caused by central and peripheral attenuation of sympathetic outflow and central activation of nonadrenergic imidazoline-preferring receptors.[176] [224] [225] [226] The decrease in central sympathetic outflow reduces activity in peripheral sympathetic neurons without
Although α2 -agonists have not been used as sole anesthetic agents in humans, these drugs reduce the anesthetic requirement and provide a more stable cardiovascular course, presumably because of their sympatholytic effect and the need for lower doses of cardioactive anesthetic.[230] [231] [232] Clonidine reduced the halothane MAC by up to 50% in a dose-dependent fashion,[233] limited by α1 -adrenoreceptor activation at higher concentrations. Data suggest that oral, intravenous, epidural, and intrathecal administration of clonidine potentiates the anesthetic action of other agents, volatile or injectable, and reduces general and regional anesthetic requirements with correspondingly fewer side effects,[230] [231] [233] [234] [235] [236] [237] [238] [239] [240] [241] [242] [243] [244] [245] [246] [247] [248] [249] [250] [251] [252] [253] [254] although perioperative ischemia was not reduced. [255] Clonidine also attenuates the rise in intraocular pressure associated with laryngoscopy and endotracheal intubation.[256] [257] [258]
Another α2 -agonist, dexmedetomidine, which has a half-life of 2.3 hours and has a 1600:1 preference for α2 -receptors relative to α1 -receptors,[259] was introduced into clinical practice as an adjunct to regional, local, and general anesthetics.[260] Dexmedetomidine decreases the halothane MAC by more than 95% in animals.[233] [261] [262] In healthy volunteers, dexmedetomidine increases sedation, analgesia, and memory loss and decreases heart rate, cardiac output, and circulating catecholamines in a dose-dependent fashion. [259] The purported MAC-reducing sedative and analgesic effects in preclinical and volunteer studies have been largely borne out in clinical practice. Dexmedetomidine infusions attenuate the hemodynamic lability of induction, maintenance, and emergence, but they require careful down titration of other agents to prevent overdosing[263] [264] because requirements for other anesthetics may decrease.[256] Another use for this drug is to provide sedation for mechanically ventilated patients during weaning from the ventilator.[265] Dexmedetomidine is administered as a loading dose of 1 µg/kg over 10 to 20 minutes followed by an infusion at 0.4 to 0.7 µg/kg/hour.
In addition to their use in the operative setting, α2 -agonists provide effective analgesia for acute and chronic pain, particularly as adjuncts to local anesthetics and opioids. The addition of clonidine increased duration of analgesia and reduced doses of each component.[240] [242] [243] [266] [267] [268] [269] [270] [271] [272] [273] [274] [275] [276] [277] [278] [279] [280] Epidural clonidine is indicated for the treatment of intractable pain, which is the basis for approval of parenteral clonidine in the United States as an orphan drug.[281] Patients with intractable pain that is unresponsive to maximum doses of oral or epidural opioids benefit from oral, patch, intramuscular, and neuraxial administration of clonidine,[282] [283] [284] [285] [286] as do patients with reflex sympathetic dystrophy[287] and neuropathic pain.[283] [288] The intrinsic analgesic effects of α2 -agonists have been demonstrated with large doses of clonidine alone, administered intrathecally (as much as 450 µg) or epidurally (1 to 2 µg/kg/hour) to control intraoperative and postoperative pain. Clonidine decreases postoperative oxygen consumption and adrenergic stress response.[289] [290] Despite dose-dependent adverse effects such as hypotension and sedation and idiosyncratic adverse effects such as bradycardia, clonidine does not induce profound respiratory depression and only mildly potentiates opiate-induced respiratory depression.[291] [292] [293]
Aside from its role as an anesthetic adjuvant and antihypertensive agent, clonidine has been used to treat panic disorder[294] ; symptoms of opiate, benzodiazepine, and ethanol withdrawal[295] [296] ; cigarette craving after smoking cessation [297] [298] [299] ; and emesis in cancer chemotherapeutic regimens. Its use in diabetic diarrheas is based on the presence of α2 -receptors on gut epithelial cells. Normally, stimulation of ileal mucosal α2 -receptors by epinephrine promotes sodium chloride absorption and inhibits bicarbonate secretion. Intractable diarrhea can be a major problem in diabetes because mucosal norepinephrine stores are depleted, and denervation hypersensitivity increases the numbers of postsynaptic α2 -receptors. Clonidine may increase blood glucose concentrations by inhibiting insulin release.[300] Unlike spinal opioids, clonidine does not cause urinary retention and may hasten the time to first micturition after spinal anesthesia.[300] [301] [302] [303]
Although at clinical doses it can act at β2 - and α1 -receptors, dobutamine, a synthetic analog of dopamine, has predominantly β1 -adrenergic effects. Compared with isoproterenol, it is reported to affect inotropy more than chronotropy, but it increases conduction velocity through nodal tissue to the same extent.[318] It exerts less β2 -type effect than isoproterenol and less α1 -type effect than norepinephrine. Unlike dopamine, it does not directly release endogenous norepinephrine, nor does it act at dopaminergic receptors.
Dobutamine is particularly useful in CHF and MI complicated by a low-output state, although in cases of severe hypotension, it may not be effective because it lacks significant α1 -pressor effect. Although dobutamine increases cardiac contractility in the failing heart, its ability to lower filling pressures contrasts with the effects of dopamine and norepinephrine. It is relatively safe to use in myocardial ischemia without increasing the size of the infarct or causing arrhythmias.[319] Doses less than 20 µg/kg/min do not produce tachycardia, but in especially severe CHF, significant tachycardia is the primary adverse effect. Because dobutamine directly stimulates β1 -receptors, it does not rely on norepinephrine stores and
The β2 -vasodilating effects of dobutamine are almost exactly offset by its α1 -constricting effects, which experimentally can be revealed by administering a nonselective β-blocking drug.[320] Its modest ability to dilate the peripheral vasculature is probably more closely connected with its ability to relieve the high-adrenergic state of decompensated CHF than to specific β2 -mediated vasodilation.[321] Clinical situations that call for distinct afterload reduction may be better served by an agent such as nitroprusside.
Prolonged treatment with dobutamine causes down-regulation of β-receptors; tolerance to its hemodynamic effects is significant after 3 days and may be temporarily offset by increasing the rate of infusion.[322] Intermittent infusions of dobutamine have been used in the long-term treatment of heart failure and have improved exercise tolerance[323] but not survival.[324]
Isoproterenol (Isuprel) provides relatively pure nonselective β-adrenergic stimulation with no significant effect at α-receptors. Its β1 -adrenergic stimulation is significantly stronger than β2 , but it still causes more β2 -adrenergic activity than dobutamine. Since the development of other inotropes, its popularity has declined because of its adverse effects of tachycardia and arrhythmias. Isoproterenol historically was used in bradycardia or heart block resistant to atropine, but it is not included in the most recent American Heart Association Advanced Cardiac Life Support protocol. Its primary use at this time is as a chronotropic agent in patients after heart transplantation. These patients are unable to generate an endogenous sympathetic response to challenges because the sympathetic fibers are divided when the native heart is removed. The availability of superior pharmacologic options for most clinical indications has led to Isuprel's removal from many hospital formularies. Infusion rates start at 0.5 to 5 µg/min for adults. Because it is not taken up into adrenergic nerve endings, its duration of action is slightly longer than that of the natural catecholamines.
In the past, isoproterenol was used in the treatment of bronchospasm for its β2 -adrenergic stimulating properties, but unpleasant and dangerous β1 -mediated adverse effects limited its use. The development of β2 -selective agents has made β-stimulants a cornerstone of the treatment of bronchospasm. However, this β2 -selectivity is only relative, and it may be lost at higher doses; in addition, β2 -receptors in the sinoatrial node may cause tachycardia when stimulated. The structures of these drugs have also been modified to slow their metabolism, prolonging therapeutic benefit and enabling oral administration. In particular, the addition of bulky structures on the catecholamine amino group increases β2 -selectivity, decreases affinity for α-receptors, and protects against metabolism by COMT. These agents are aerosolized and given by inhaler for rapid onset and to minimize systemic drug levels and adverse effects.
An increase in the annual number of deaths from asthma has been well documented, and it has been suggested that this increase may be related to β2 -agonist use.[325] [326] [327] A susceptibility to arrhythmia caused by these agents by direct cardiac stimulation or by β2 -induced hypokalemia is one suggested mechanism. It has also been hypothesized that the long-term use of these drugs may increase airway hyperreactivity. Nonetheless, their safe use in many thousands of patients is well documented.
Commonly used drugs include metaproterenol (Alupent, Metaprel), terbutaline (Brethine, Bricanyl), and albuterol (Proventil, Ventolin). Metaproterenol is probably less β2 -selective than albuterol or terbutaline. Terbutaline is the only β2 -selective agent that can be given subcutaneously and therefore may have particular use in status asthmaticus. The normal subcutaneous dose is 0.25 mg, which may be repeated after 15 to 30 minutes.
The β2 -agonists are also used to treat premature labor.[328] Ritodrine (Yutopar) has been marketed for this purpose. β1 -Adrenergic adverse effects are common, particularly when the drugs are used intravenously. The other β2 -selective drugs have also been used as tocolytics, and all have been associated with significant β1 -adrenergic adverse effects and the occasional incidence of pulmonary edema.[329] Their use for this purpose has recently been questioned.[330]
α1 -Antagonists have long been used clinically as antihypertensives, but they have become less popular over the years. An α1 -blockade vasodilates by blocking the effect of endogenous catecholamines on arterial and venous constriction. The effects are potentiated when standing or in the presence of hypovolemia. Reflex tachycardia and fluid retention can ensue. The α2 -antagonists may act presynaptically to release norepinephrine.
Phenoxybenzamine (Dibenzyline) is the prototypical α1 -antagonist, although it irreversibly binds to α1 - and α2 -receptors. New receptors must be synthesized before complete offset of its effects occurs. Its half-life after oral administration is unknown, but that after an intravenous dose is about 24 hours. Phenoxybenzamine decreases peripheral resistance and increases cardiac output; blood flow to skin and viscera is increased. As expected, its primary adverse effect is orthostatic hypotension; nasal stuffiness may occur. In addition to receptor blockade, phenoxybenzamine inhibits neuronal and extraneuronal uptake of the catecholamines. Phenoxybenzamine is used in the treatment of pheochromocytoma; with extended use, it establishes a "chemical sympathectomy" preoperatively that aids in blood pressure control, permits correction of the contracted plasma volume, and protects against catecholamine-induced cardiac damage. Phenoxybenzamine treatment allows for a smoother perioperative course. The total daily dose is 40 to 120 mg in two or three divided doses. When exogenous sympathomimetics are administered after α1 -receptor blockade, their vasoconstrictive effects are inhibited. The effect of phenylephrine is completely blocked, whereas that of norepinephrine is limited to its β1 -adrenergic effect of cardiac stimulation. Potential epinephrine reversal caused by unopposed β2 -agonism manifests as severe
Phentolamine (Regitine) is a shorter-acting drug that blocks α1 - and α2 -receptors. Historically used to treat pulmonary hypertension, it has been largely supplanted by nitroglycerin and nitroprusside. It is also used to treat hypertension associated with clonidine withdrawal or with tyramine ingestion during MAOI therapy, but few data have been collected on its efficacy and safety for these indications. The dose is 1 to 5 mg of phentolamine given intramuscularly or slowly intravenously. Its plasma half-life is 19 minutes after intravenous administration. Phentolamine has also been infiltrated into affected tissues after extravasation of agents such as norepinephrine in an attempt to relax vasoconstriction; for this purpose, 5 to 10 mg is diluted in 10 mL of saline. Adverse effects of phentolamine include hypotension and gastrointestinal distress; reflex tachycardia and arrhythmias may result from action at α2 -receptors. Coronary artery disease and peptic ulcer disease are relative contraindications. As in phenoxybenzamine overdosage, severe hypotension may require treatment with norepinephrine rather than epinephrine. Tolazoline (Priscoline) is a related drug used in persistent pulmonary hypertension of the newborn.[331]
Prazosin (Minipress) is a potent selective α1 -adrenergic blocker often used as a prototypic antagonist in pharmacologic experiments. It antagonizes the vasoconstrictor effects of norepinephrine and epinephrine, causing a decline in peripheral vascular resistance and venous return to the heart. Although heart rate does not normally increase, orthostatic hypotension is a major problem. Unlike other antihypertensive drugs, prazosin improves lipid profiles, lowering low-density lipid levels while raising the level of high-density lipids. It is primarily used to treat hypertension. It has also been used in CHF, but unlike the angiotensin-converting enzyme inhibitors (ACEIs), prazosin does not prolong life. It is metabolized by the liver. Supplied as 1-, 2-, and 5-mg tablets, its starting dose is usually 0.5 to 1 mg, given at bedtime because of the orthostatic hypotension. Eventually, it can be given twice daily.[331]
α2 -Antagonists, such as yohimbine, increase sympathetic outflow by enhancing release of norepinephrine. These drugs have proved to be of little clinical utility in anesthesia, although they are used in urology.
The β-adrenergic receptor antagonists (i.e., β-blockers) are among the most commonly prescribed drugs and are frequently taken by patients presenting for surgery. Current indications for the use of β-blockade include ischemic heart disease, postinfarction management, arrhythmias, hypertrophic cardiomyopathy, hypertension, heart failure, and prophylaxis for migraine headache. Concern that patients treated with β-blockers would be hemodynamically unstable under anesthesia has proved to be largely unjustified. These drugs are an important part of the armamentarium of the anesthesiologist in the ongoing attempt to limit stress responses perioperatively and to protect the cardiovascular system. A comprehensive analysis by the Agency for Healthcare Research and Quality found that use of β-blockers perioperatively reduced the morbidity and mortality in noncardiac surgery for patients at risk.[332] Several well-known clinical trials[333] [334] [335] [336] [337] and the widespread use and safety of these drugs in heart failure[338] have made β-blockade an evolving standard.
A wide spectrum of β-adrenergic blockers is available to the clinician. The most important properties to consider when choosing a β-blocker for long-term use are cardioselectivity, intrinsic sympathomimetic activity, and lipid solubility.[339] [340] In anesthetic practice, cardioselectivity, duration of action, and a formulation suitable for intravenous use are crucial factors ( Table 16-11 ). The β-antagonists resemble isoproterenol in structure and bind competitively to the β-receptor, blocking access by more potent β-agonists ( Fig. 16-21 ).[331] Competitive inhibition at the β-receptor can be overcome by increasing the available concentration of β-agonist. The potency of a β-blocker is often determined by its ability to inhibit induction of tachycardia by isoproterenol. Propranolol is assigned a potency of 1, and the other drugs are evaluated in relation to it.
Nonselective β-adrenergic blockers act at the β1 - and β2 -receptors. The nonselective β-blockers include propranolol, nadolol, pindolol, sotalol, oxprenolol, penbutolol, and timolol.[321] [340] Cardioselective β-blockers have a stronger affinity for β1 -adrenergic receptors than for β2 -adrenergic receptors, and the predominant effects therefore are cardiac. Velocity of atrioventricular conduction, heart rate, and cardiac contractility are decreased, as is the release of renin by the juxtaglomerular apparatus and lipolysis at the adipocytes. With larger doses, the relative selectivity for β1 -adrenergic receptors is lost, and β2 -receptors are also blocked,[341] with potential bronchoconstriction, peripheral vasoconstriction, and decreased glycogenolysis. Cardioselective drugs include atenolol, betaxolol, bevantolol, esmolol, and metoprolol. [321] [340] These drugs may be preferable for patients with obstructive pulmonary disease, peripheral vascular disease, Raynaud's phenomenon, and diabetes mellitus. Although previously a matter of controversy, a meta-analysis concluded that cardioselective β-blockers can be given safely to patients with chronic obstructive pulmonary disease.[342] Nevertheless, because the selectivity is only relative and may be lost at conventional clinical doses, extreme care should be used in administering a β-blocker in the presence of pulmonary disease. Some β-antagonists also have vasodilatory effects, making them particularly useful in the treatment of hypertension[321] and CHF.[343] [344] Labetalol vasodilates by blocking α1 -receptors and by direct β2 -receptor agonism.
β-Blockers exerting a partial agonist effect at the receptor while blocking access by more potent agonists possess intrinsic sympathomimetic activity (ISA). Agents with ISA include acebutolol, carteolol, celiprolol, dilevalol, oxprenolol, penbutolol, and pindolol.[340] Of these, acebutolol and pindolol are most often used in the United States, but neither is commonly used in anesthetic practice. These agents lower blood pressure with less decrease in resting heart rate and left ventricular function.
| Characteristic | Atenolol | Metoprolol | Propranolol HCl | Labetalol | Esmolol | Carvedilol |
|---|---|---|---|---|---|---|
| Proprietary name | Tenormin | Lopressor | Inderal Ipran | Trandate | Brevibloc | Coreg |
|
|
|
|
|
Normodye |
|
|
| Relative β sensitivity | + | + | 0 | 0 | + | 0 |
| Intrinsic sympathetic activity | 0 | 0 | 0 | + | 0 | 0 |
| Membrane-stabilizing activity | 0 | 0 | ++ | 0 | 0 | — * |
| Lipophilicity † | Low | Moderate | High | Low | Low | High |
| Predominant route of elimination | RE (mostly unchanged) | HM | HM | HM | Hydrolysis by RBC esterase | HM |
| Drug accumulation in renal disease | Yes | No | No | No | No | No |
| Elimination half-life (hr) | 6 to 9 | 3 to 4 | 3 to 4 | ≅6 | 9 min | 2 to 8 |
| Usual maintenance dose PO | 50–100 mg qd | 50–100 mg qid | 60 mg qid | 100–600 mg bid | N/A | 25–50 mg bid |
| Usual IV dose (caution) |
|
5 mg q 5 min × 3 | 0.1 mg/kg (maximum) | 1–2 mg/kg | 50–300 µg/kg/min infusion | 15 mg |
| HM, hepatic metabolism; N/A, not applicable; RE, renal excretion; 0, no effect; +, mild effect; ++, moderate effect. | ||||||
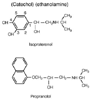 Figure 16-21
Structures of isoproterenol and propranolol. (From
Tollenaeré JP: Atlas of the Three-Dimensional Structure of Drugs. Amsterdam,
Elsevier North-Holland, 1979.)
Figure 16-21
Structures of isoproterenol and propranolol. (From
Tollenaeré JP: Atlas of the Three-Dimensional Structure of Drugs. Amsterdam,
Elsevier North-Holland, 1979.)
The degree of lipid solubility greatly affects absorption and metabolism. Propranolol, metoprolol, and pindolol, the most lipid-soluble agents, are readily absorbed from the gastrointestinal tract and metabolized in the liver, undergoing extensive first-pass metabolism. Propranolol and metoprolol are given in a much lower dose intravenously than orally because up to 70% of the orally administered drug is removed on the first pass of portal blood through the liver.[339] Altered hepatic function may have a profound effect on plasma levels. The oral or intravenous dose of these agents required to reach a clinical end point varies greatly between patients. Because of a relatively short half-life, lipid-soluble agents are usually given at least twice each day.[340] The extremely short half-life of esmolol, however, results from its metabolism by esterases.
The non-lipid-soluble, or hydrophilic, drugs are atenolol, sotalol, and nadolol. They are not as readily absorbed from the gastrointestinal tract or as extensively metabolized, and they are excreted unchanged by the kidneys. Because of a relatively long half-life, daily dosing may be adequate. There is less interpatient variability in plasma drug levels and clinical effects because of the lack of hepatic metabolism and first-pass effect.[340] Because the lipid-insoluble drugs are slower to cross the blood-brain barrier, they may avoid the CNS adverse effects associated
Propranolol and acebutolol possess membrane-stabilizing activity (MSA), also referred to as the quinidine-like or local anesthetic effect. This effect reduces the rate of rise of the cardiac action potential. The MSA of propranolol may explain the decrease in the oxygen affinity of hemoglobin[351] when it is administered.[352] However, MSA is seen only at concentrations 10 times that required for blockade of β-receptors[353] and is probably of little clinical consequence. Overdose with agents with MSA is associated with a higher incidence of fatality.[354]
β-Adrenergic antagonists have been used to establish perioperative β-blockade (see Chapter 25 ). Compelling results from the multicenter study of a perioperative ischemia research group[330] have shown that β-blockade improves outcomes. Atenolol was given before and after surgery and continued for the duration of the hospitalization. In this study, patients were randomly allocated to receive atenolol or placebo preoperatively, intraoperatively, and until discharge; follow-up lasted more than 2 years. The overall mortality rate after discharge from the hospital was significantly less for the treated group. Surprisingly, this difference in survival persisted at the 2-year point (68% survival in the placebo group and 83% in the atenolol group). A meta-analysis of this and subsequent studies has confirmed that β-antagonists have a significant role in preventing perioperative cardiac morbidity, particularly in patients at risk for cardiac events.[355]
The safety of continuing β-blockade perioperatively is well established, and initial concerns regarding interaction with general anesthesia have not beenconfirmed.[356] Attempts to discontinue β-blockers increase the risk of rebound tachycardia (with or without atrial fibrillation) and myocardial ischemia in patients with coronary disease. These drugs should be given up to the time of surgery, and intravenous forms in appropriate dosages should be used whenever gastrointestinal absorption may be in question. Doses for intravenous administration vary less among patients because of the absence of first-pass hepatic effects. If β-blockers have been omitted from the preoperative regimen, esmolol or labetalol may be used acutely to blunt tachycardia and hypertension. Cardioselective and nonselective β-blockers appear effective in blocking chronotropic effects of endotracheal intubation and surgical stress.[357]
Propranolol was initially introduced for treatment of myocardial ischemia 3 decades ago, and β-blocking drugs remain an important part of drug therapy for myocardial ischemia (see Chapter 50 ). This class of drugs reduces oxygen demand by decreasing heart rate and cardiac contractility. Cardioselective and nonselective β-blockers are effective. Atenolol, metoprolol, nadolo, and propranolol have been approved in the United States for the treatment of angina; metoprolol and atenolol are the only β-blockers approved for intravenous use in acute MI.[340] Although initially there was concern that β2 -receptor blockade could worsen ischemia through unopposed α-mediated vasoconstriction, this phenomenon is rarely seen, even in patients with variant angina. In usual clinical practice, the dose is increased until the heart rate is 60 to 80 beats/min at rest and until there is no tachycardia with exercise. In theory, a nonselective antagonist may seem a better choice in acute MI because the blockade of β2 -receptors may protect against stress-induced potassium shifts and hypokalemia-associated arrhythmias, but cardioselective and nonselective drugs appear equally useful.[321] Agents with ISA are not as beneficial in this situation.[321] The β-antagonists are used in treating acute MI and on a long-term basis in patients after infarction to reduce reinfarction and mortality.[358] [359] [360] [361] Early administration of intravenous β-blocking agents to patients receiving thrombolytic therapy appears to lower the incidence of ischemia and reinfarction [362] and may reduce the incidence of serious ventricular arrhythmias.[363] The long-term use of β-blockers (i.e., timolol, propranolol, metoprolol, and atenolol) has been shown unequivocally to decrease mortality after MI.
Over the past 5 years, β-blockers have become first-line agents for the treatment of CHF of ischemic and nonischemic origin. Clinicians had been disinclined to use β-blockers for heart failure patients because of the drugs' negative chronotropic and inotropic effects. These effects have not turned out to be significant concerns in clinical practice. Early studies demonstrated a significant decrease in all-cause mortality for heart failure patients treated with metoprolol[343] or bisoprolol.[344] Several large studies[364] [365] were stopped because the decrease in mortality in treated patients with moderate to severe heart failure was so pronounced. The benefits of β-blockers in patients with heart failure have been attributed to normalization and remodeling of the ventricle that begins after 1 month of β-blockade,[366] to decreased norepinephrine-related cardiomyocyte apoptosis in the presence of β1 -blockade, [367] and to decreased sudden cardiac death because of the β-blockers' antiarrhythmic activity. To avoid worsening a patient's heart failure symptoms and to minimize negative inotropic effects, β-blockers are started at very low doses and titrated to the target dose.[368] The only β-blocker approved for treatment of CHF by the U.S. Food and Drug Administration is carvedilol, and its efficacy in patients with severe (i.e., New York Heart Association class IV) heart failure is still being studied. If decompensation occurs with the β-blocker, the phosphodiesterase inhibitors are the inotropes of choice because their effects are not antagonized by β-blockade.[369]
Although β-blockers are recognized by the Joint National Council of the USA Guidelines as one of the first-line therapies for hypertension, [370] the mechanisms by which β-antagonists treat hypertension are incompletely understood. Blood pressure reduction is specific to hypertensive patients because long-term treatment of normotensive individuals does not lower blood pressure. Reduced cardiac output and renin release have been suggested as mechanisms. In hypertensive patients, β-antagonists without ISA may cause a 15% to 20% reduction in cardiac output and a 60% reduction in renin release. However, pindolol, which has ISA and minimal
The β-blockers are widely used in the therapy of tachyarrhythmias as class II agents (see Chapter 32 ). Two possible mechanisms of action are blockade of catecholamine effects and MSA, although the latter is most likely not clinically significant because antiarrhythmic effects are present in agents without MSA.[374] β-Antagonists slow the rate of depolarization of the sinus node and any ectopic pacemakers, slow conduction through atrial tissue and the atrioventricular node, and increase the refractory period of the atrioventricular node. These drugs can convert atrial arrhythmias to sinus rhythm,[375] but β-blockade is primarily used to slow the ventricular response. Reentrant tachyarrhythmias and those associated with Wolff-Parkinson-White syndrome, mitral valve prolapse, and prolonged QT interval may also respond to these drugs.[353] Care should be exercised if an atrioventricular block is present, as in digitalis toxicity, although these drugs are useful in the treatment of digitalis-associated tachyarrhythmias.[353] Sotalol, a β-blocker with added class III activity, is effective against ventricular tachyarrhythmias; however, in a trial comparing racemic sotalol with D-sotalol in post-MI patients to protect against ventricular fibrillation, mortality increased with D-sotalol.[376] The trial was terminated when patients at lowest risk for arrhythmic events after MI had an increased mortality rate with this drug.[377]
β-Antagonists are frequently used as adjuvants to moderate the reflex tachycardia associated with vasodilators. This tachycardia can limit the effectiveness of blood pressure control or may cause myocardial ischemia. It is crucial that appropriate β-blockade be administered during vasodilator therapy of aortic dissection. In addition to potentiating blood pressure reduction, β-blockade also reduces the velocity of left ventricular ejection (dp/dt) to attenuate the shearing force associated with the increased velocity of ventricular contraction when nitroprusside is used without concomitant β-blockade.[378] Labetalol has been particularly useful in this situation.[379]
Cardiac complications are a primary cause of morbidity in thyrotoxicosis (see Chapter 27 ). β-Blockade can suppress the tachycardia and rhythm disturbances, although very large doses may be required. β-Antagonists also may be combined with digitalis for their synergistic effect on atrioventricular node conduction. Propranolol inhibits conversion of thyroxine to the active form, triiodothyronine, in the periphery.[380]
Timolol (Timoptic) and betaxolol (Betoptic) are β-blocking drugs used topically in the eye to treat glaucoma. They reduce the production of aqueous humor. Even topical use of these agents has been associated with significant systemic effects of β-blockade. The β-blockers are used in idiopathic hypertrophic subaortic stenosis to reduce the dynamic obstruction to left ventricular outflow. The drugs are also effective in prophylaxis, but not therapy, of migraine headaches and in controlling acute panic symptoms and essential tremor.
The adverse effects of most concern are those involving cardiopulmonary function. Severe noncardiopulmonary reactions such as cutaneous reactions or anaphylaxis are rare. Life-threatening bradycardia, even asystole, may occur, and decreased contractility may precipitate CHF in vulnerable individuals. In patients with bronchospastic lung disease, β2 -blockade may be fatal. CNS effects, although an appropriate consideration in long-term therapy, are not a concern in the usual anesthetic use of these agents. Diabetes mellitus is a relative contraindication to the long-term use of β-antagonists, because hypoglycemia in the face of sympathetic blockade is not accompanied by warning signs such as tachycardia and tremor, and compensatory glycogenolysis is blunted. However, most non-insulin-dependent diabetic patients can tolerate these drugs, although β-blockade may cause insulin resistance in rare cases. In addition to the potential worsening of peripheral perfusion by β2 -blockade in patients with peripheral vascular disease, the Raynaud phenomenon may be triggered in susceptible patients. Sudden withdrawal of β-blockers may cause myocardial ischemia and possibly infarction, although this is less of a problem with β-blockers with ISA such as pindolol.[368] [381] [382] Although β-antagonists may reduce renal blood flow and glomerular filtration rate, these agents can be used in renal failure. For these patients, the doses of lipid-insoluble drugs should be reduced. To avoid worsening of hypertension, use in pheochromocytoma should be avoided unless α-receptors have previously been blocked. Nonselective agents may elicit hypertensive responses in cases of high sympathetic stimulation.[383]
Undesirable drug interactions are possible with β-blockers. [331] [340] The rate and contractility effects of verapamil are additive to those of the β-blockers, [384] [385] and care should be taken in combining these agents, especially the intravenous forms in acute situations such as supraventricular tachycardia. The combination of digoxin and β-blockers can have powerful effects on heart rate and conduction and should be used with special care. Pharmacokinetic interactions are predictable from the degree of lipid solubility of the drug. Cimetidine and hydralazine may reduce hepatic perfusion, thereby increasing plasma levels and half-lives of the lipid-soluble β-antagonists. Barbiturates, phenytoin, rifampin, and smoking may induce hepatic enzymes, enhancing metabolism. Propranolol may reduce hepatic clearance of lidocaine, increasing the risk of toxicity.
Overdose of β-blocking drugs may be treated with atropine, but isoproterenol, dobutamine, or glucagon infusions (or some combination) may be required along
The drugs propranolol, metoprolol, labetalol, and esmolol are particularly useful in anesthetic practice because they are widely available in intravenous formulation and have well-characterized effects. If the drug the patient has taken on a long-term basis is propranolol, metoprolol, or labetalol, it may be continued in intravenous form if the clinical situation is relatively stable. In deciding which intravenous β-blocker to substitute in a patient taking a β-blocker on a long-term basis, the need for cardioselectivity is a primary consideration. Cardioselectivity is provided by metoprolol or esmolol. If the long-term agent has ISA, oxprenolol and acebutolol have intravenous forms, but they are not readily available. In many situations, esmolol may be substituted and titrated to effect, with the expectation that its effects will fade relatively rapidly in case it is not well tolerated.
Propranolol (Inderal, Ipran) is the prototypic β-blocker, and its actions and its use are well characterized.[374] It is a nonselective β-blocking drug with MSA but no ISA.[340] It readily penetrates the CNS. Because of its high lipid solubility, it is extensively metabolized in the liver, but metabolism varies greatly from patient to patient. Its effective dose is extremely variable: 10 mg to as much as 320 mg may be given orally each day. Clearance of the drug can be affected by liver disease or altered hepatic blood flow. Renal impairment does not require adjustment of dosing. Despite its half-life of 4 hours, its antihypertensive effect persists long enough to permit dosing once or twice daily.[374] [386] Inderal LA is a sustained-release formulation given once daily.[331]
Propranolol is available in an intravenous form; although initially used as bolus or infusion, the infusion has been largely supplanted by esmolol. For bolus administration, doses of 0.1 mg/kg may be given, although most practitioners initiate therapy with much smaller doses, typically 0.25 to 0.5 mg, and titrate to effect. Propranolol shifts the oxyhemoglobin dissociation curve to the right, perhaps accounting for its efficacy in vasospastic disorders.[387] [388] [389]
Metoprolol (Lopressor) is approved for the treatment of angina pectoris and is the only intravenous β-blocker formally approved by the U.S. Food and Drug Administration for treatment of acute MI. Lacking ISA and MSA, it is cardioselective. Because it is metabolized in the liver by the monooxygenase system, doses need not be adjusted in the presence of renal failure.[340] The usual oral dose is 100 to 200 mg/day, once or twice daily for hypertension and twice daily for angina pectoris. It may be administered intravenously in doses of 2.5 to 5 mg every 2 to 5 minutes up to about 15 mg, titrating to heart rate and blood pressure.
Labetalol (Trandate, Normodyne) is representative of a class of drugs that act as competitive antagonists at the α1 - and β-adrenergic receptors. Labetalol consists of four isomers that block the α1 -, β1 -, and β2 -receptors; inhibit neuronal uptake of norepinephrine (uptake-1); act as a partial agonist at β2 -receptors; and possibly have some direct dilating abilities.[331] The potency of the mixture for β-blockade is 5- to 10-fold that for α-blockade. [390] The usual oral dose of labetalol is 200 to 400 mg twice daily, although much larger doses have been used. It is metabolized by the liver, and clearance is affected by hepatic perfusion. The dose need not be adjusted for renal dysfunction.[340] Labetalol may be given intravenously every 5 minutes in 5- to 10-mg doses or up to a 2-mg/min infusion. It significantly blunts cardiovascular responses to tracheal intubation. [391] It can be effective in treatment of aortic dissection,[379] hypertensive emergencies,[392] [393] and postoperative cardiac surgical patients, [394] particularly because vasodilation is not accompanied by tachycardia. It may be used in pregnancy to treat hypertension on a long-term basis and in more urgent situations.[395] Uterine blood flow is not affected even with a significant reduction of blood pressure.[396]
Carvedilol, a mixed α- and β-antagonist, has been introduced as therapy for mild or moderate hypertension,[397] [398] [399] [400] [401] [402] [403] [404] [405] for management of stable or unstable angina, and after acute MI.[406] [407] [408] [409] [410] Clinical trials of carvedilol use in the treatment of controlled CHF (New York Heart Association class II-IV) suggest a significant reduction in mortality,[364] [365] [411] [412] especially for patients with diabetes.[413]
Because of its hydrolysis by esterases, esmolol (Brevibloc) has a uniquely short half-life of 9 to 10 minutes, which makes it particularly useful in anesthetic practice. It is administered when β-blockade of short duration is desired or in critically ill patients for whom adverse effects of bradycardia, heart failure, or hypotension may necessitate rapid withdrawal of the drug. Peak effects of a loading dose are seen within 5 to 10 minutes and diminish rapidly (within 20 to 30 minutes). It is cardioselective. It may be given as a bolus of 0.5 mg/kg to blunt cardiovascular responses to tracheal intubation. If used as an infusion for the treatment of supraventricular tachycardia, 500 µg/kg is given over 1 minute, followed by an infusion of 50 µg/kg/min for 4 minutes. If the rate is not controlled, a repeat loading dose followed by a 4-minute infusion of 100 µg/kg/min is given, and this sequence is repeated, increasing the infusion in 50-µg/kg/min increments, up to 200 or 300 µg/kg/min if needed. The effect may persist for 20 to 30 minutes after discontinuation of the infusion. Compared with verapamil, esmolol is more likely to convert atrial fibrillation to sinus rhythm.[375] Esmolol is safe and effective in treatment of intraoperative and postoperative hypertension and tachycardia.[414] [415] [416] If continuous use is required, it may be reasonably replaced by a longer-lasting cardioselective drug such as intravenous metoprolol. It has been used safely even in patients with compromised left ventricular function. [417] [418]
Some early antihypertensive drugs acted by replacing norepinephrine in the nerve ending with a much less potent false transmitter. Methyldopa (Aldomet) is such a drug and was the most popular nondiuretic antihypertensive used before the development of β-blockers.[339] Like DOPA, methyldopa enters the biosynthetic pathway for norepinephrine (see Fig. 16-9 ). It is then decarboxylated to
Methylparatyrosine (metyrosine, Demser) is a potent inhibitor of tyrosine hydroxylase, which catalyzes the formation of DOPA from tyrosine (see Fig. 16-9 ). Because this is the rate-limiting step in the biosynthesis of norepinephrine, the drug significantly decreases levels of endogenous catecholamines and is useful in treating inoperable or malignant pheochromocytomas.[421]
Reserpine affects the uptake of norepinephrine not at the neuronal membrane but at the vesicular membrane, thereby inhibiting transport and storage of norepinephrine and dopamine. Eventually, norepinephrine stores are depleted, and postsynaptic receptors increase in number, which may increase the effects of epinephrine in patients who have been given reserpine (see Table 16-5 ). When patients who take reserpine require a sympathomimetic drug, direct-acting agents are more useful than mixed-acting drugs, which are efficacious only at higher doses, if at all. Reserpine has central effects that lead to sedation and depression; peripheral effects are responsible for its utility as an antihypertensive drug.
Guanethidine (Ismelin) acts initially by blocking the release of norepinephrine, and then it is taken up into adrenergic nerve endings by the uptake-1 mechanism and depletes norepinephrine stores. It is used to treat hypertension, usually after many other drugs have been tried. Its inability to cross the blood-brain barrier accounts for its lack of sedative effects. Guanadrel (Hylorel) is similar to guanethidine, but it has a faster onset and shorter duration of action.
Bretylium, a class III antiarrhythmic used parenterally to treat life-threatening ventricular tachyarrhythmias, is now largely of historical interest. Like guanethidine, it is taken up into adrenergic nerve terminals, but its mechanism of action is otherwise quite different. Bretylium initially causes norepinephrine release and subsequently blocks that release by decreasing sympathetic nerve excitability. Unlike guanethidine, bretylium does not deplete norepinephrine stores.[422] The initial catecholamine release may worsen some arrhythmias, such as those associated with digitalis toxicity and myocardial ischemia.[353] As a consequence, bretylium, which had been part of the advanced cardiac life support protocols for treating ventricular arrhythmias, has been dropped from the algorithms.
MAO and COMT are enzymes important in degradation of the catecholamines. MAOIs bind irreversibly to the enzyme and increase amine concentration within the presynaptic terminal. This increase is associated with antihypertensive, antidepressant, and antinarcoleptic effects.[423] MAOIs are thought to exert their antihypertensive effect through a false transmitter mechanism. Tyramine is usually oxidatively deaminated in the gut by MAO. With administration of an MAOI, tyramine levels rise. When tyramine is taken up into the sympathetic nerve terminal by the uptake-1 mechanism, it enters the varicosities and is transformed by DBH into octopamine. On its subsequent release in place of norepinephrine, octopamine is only weakly reactive at sympathetic receptors, resulting in a lowering of blood pressure. The MAOIs are no longer used as antihypertensives because many other drugs with better risk-benefit profiles have been developed.
MAOIs are primarily used in psychiatric practice. The use of MAOIs as antidepressants is based on the theory that depression is caused by decreased amine in the synapses of the CNS. Inhibition of MAO makes more amine available for release. MAOIs available for treatment of depression include isocarboxazid (Marplan), phenelzine sulfate (Nardil), and tranylcypromine sulfate (Parnate).
Based on substrate specificity, there are at least two forms of MAO. MAO-A acts on 5-HT, norepinephrine, and dopamine, whereas MAO-B is specific for tyramine. A specific MAO-B inhibitor, selegiline hydrochloride (deprenyl), has been developed for the treatment of Parkinson's disease, in hope that by blocking central dopamine breakdown, more dopamine will be preserved in the affected areas. [424]
Drug and food reactions have been of great concern in patients taking MAOIs. Tyramine-containing foods such as red wine and aged cheese must be avoided by patients taking these drugs. Consumption delivers a huge amount of tyramine to the adrenergic nerve terminal, with a subsequent massive release of norepinephrine. This release is manifest clinically as a hypertensive crisis with the potential for MI, cerebral hemorrhage, and death. Any intake of biogenic amine precursors can be expected to increase catecholamine levels greatly, as seen with the concurrent administration of levodopa with MAOIs. The effects of sympathomimetic amines, particularly the indirectly acting drugs, are enhanced.[423] Narcotics, particularly meperidine, have been associated with hyperpyrexic coma and death in patients treated with MAOIs. The depressant effects of agents such as sedatives, alcohol, and general anesthetics are enhanced in these patients. Interactions between MAOI and tricyclic antidepressants can be disastrous.[423] Anesthetic interactions with deprenyl have not been reported, but experience with this drug is limited.[424] Great concern has been expressed that patients receiving long-term therapy with an MAOI risk life-threatening drug interactions during anesthesia. Emergency surgery in patients given an MAOI can be punctuated by marked hemodynamic instability. Severe reactions to narcotics and indirect-acting sympathomimetics, as well as altered metabolism of endogenous and exogenous catecholamines, make these patients potentially difficult to manage. Because of the possibly dangerous interactions of many drugs with MAOIs, controversy exists about the best way to anesthetize these patients.[425] [426] It has been suggested that the level of concern expressed over the years may be excessive. Although prudence and custom dictate discontinuation of an MAOI at least 2 weeks in advance of elective procedures, there appear to be rational anesthetic choices based on pharmacology and risk-benefit considerations for patients when surgery cannot be delayed.
|
|
|
|
|
|
|
|
|
|
|
|
|