Neurophysiologic Aspects of Phasic Inhibition
Different fiber types in the nerve are affected differently during
local anesthesia. At least part of this difference arises from pharmacokinetic factors.
At the onset and during recovery from clinical block, in particular, longitudinal
and radial diffusion of drug will produce concentration variations within and along
the nerve. This variation is superimposed on the dynamic use-dependent inhibition
to provide variable propagation that depends on a fiber's geometry, position within
the nerve, and functional as well as electrophysiologic properties.
Different fiber types are also differentially sensitive to local
anesthetic blockade. In vivo experiments involving equilibration of local anesthetics
by continuous superfusion of peripheral nerves, as well as experiments using percutaneous
bolus injection analogous to clinical peripheral nerve block, show unequivocally
that small myelinated axons (Aγ motor and Aδ sensory fibers) are the
most susceptible to impulse annihilation. Next in order of block are the large myelinated
(Aα and Aβ) fibers, and the least susceptible are the small, nonmyelinated
C fibers. In fact, among this last group, impulses in the slowest conducting population
(conduction velocity = 0.5 to 0.8 msec-1
) are the most resistant to local
anesthetic.[26]
The generalized notion "that local
anesthetics block the smallest fibers first or most" is clearly wrong.
Selective Susceptibility of Na+
Channel
Isoforms
Ten different Na+
channels have been physiologically
identified and biochemically sequenced. At least four of them are found in peripheral
neurons, some exclusively associated with nociceptive afferents. Obviously, it would
be clinically advantageous to selectively inhibit these channels and thus prevent
or reduce pain while sparing other functions. Unfortunately, as of this writing
(2003), no such selective compounds have been reported, either because the local
anesthetic pharmacophore is too similar among the different channel isoforms or because
the same drug features that favor inhibition of "therapeutic" target channels also
result in blockade of "toxic" target channels (e.g. cardiac Na+
channels).
On the other hand, the aberrant impulses that are often considered
the hallmark of various diseases of excitable membranes, such as abnormal repetitive
firing in neuropathic pain from sites of peripheral nerve injury, are unusually susceptible
and are abolished by systemic lidocaine doses that do not block normal propagating
impulses. The conditions for such sensitivity to these local anesthetics (i.e.,
lidocaine) appear to result from the
 Figure 14-9
Structural features of the Na+
channel that
determine local anesthetic (LA) interactions. A,
Consensus arrangement of the single peptide of the Na+
channel α-subunit
in a plasma membrane. Four domains with homologous sequences (D-1 through D-4) each
contain six α-helical segments that span the membrane (S1 to S6). Each domain
folds within itself to form one cylindrical bundle of segments, and these bundles
converge to form the functional channel's quaternary structure (B).
Activation gating that leads to channel opening results from primary movement of
the positively charged S4 segments in response to membrane depolarization (see panel
C). Fast inactivation of the channel follows binding
to the cytoplasmic end of the channel of part of the small loop that connects D-3
to D-4. Ions travel through an open channel along a pore defined at its narrowest
dimension by the P region formed by partial membrane penetration of the four extracellular
loops of protein connecting S5 and S6 in each domain. Intentional, directed mutations
of different amino acids on the channel indicate residues that are involved in LA
binding in the inner vestibule of the channel (X,
on S6 segments) and at the interior regions of the ion-discriminating "selectivity
filter (
Figure 14-9
Structural features of the Na+
channel that
determine local anesthetic (LA) interactions. A,
Consensus arrangement of the single peptide of the Na+
channel α-subunit
in a plasma membrane. Four domains with homologous sequences (D-1 through D-4) each
contain six α-helical segments that span the membrane (S1 to S6). Each domain
folds within itself to form one cylindrical bundle of segments, and these bundles
converge to form the functional channel's quaternary structure (B).
Activation gating that leads to channel opening results from primary movement of
the positively charged S4 segments in response to membrane depolarization (see panel
C). Fast inactivation of the channel follows binding
to the cytoplasmic end of the channel of part of the small loop that connects D-3
to D-4. Ions travel through an open channel along a pore defined at its narrowest
dimension by the P region formed by partial membrane penetration of the four extracellular
loops of protein connecting S5 and S6 in each domain. Intentional, directed mutations
of different amino acids on the channel indicate residues that are involved in LA
binding in the inner vestibule of the channel (X,
on S6 segments) and at the interior regions of the ion-discriminating "selectivity
filter (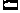 , square, on the P region), and they are also known to influence stereoselectivity
for phasic inhibition (O, also on S6 segments).
C, Schematic cross section of the channel speculating
on the manner in which S6 segments, which form a "gate," may realign during activation
to open the channel and allow entry and departure of a bupivacaine molecule by the
"hydrophilic" pathway. The closed (inactivated) channel has a more intimate association
with the LA molecule, whose favored pathway for dissociation is no longer between
S6 segments (the former pore), but now, much more slowly, laterally between segments
and then through the membrane, the "hydrophobic" pathway. Na+
ions entering
the pore will compete with the LA for a site in the channel, and H+
ions,
which pass very slowly through the pore, can enter and leave from the extracellular
opening, thereby protonating and deprotonating a bound LA molecule and thus regulating
its rate of dissociation from the channel.
, square, on the P region), and they are also known to influence stereoselectivity
for phasic inhibition (O, also on S6 segments).
C, Schematic cross section of the channel speculating
on the manner in which S6 segments, which form a "gate," may realign during activation
to open the channel and allow entry and departure of a bupivacaine molecule by the
"hydrophilic" pathway. The closed (inactivated) channel has a more intimate association
with the LA molecule, whose favored pathway for dissociation is no longer between
S6 segments (the former pore), but now, much more slowly, laterally between segments
and then through the membrane, the "hydrophobic" pathway. Na+
ions entering
the pore will compete with the LA for a site in the channel, and H+
ions,
which pass very slowly through the pore, can enter and leave from the extracellular
opening, thereby protonating and deprotonating a bound LA molecule and thus regulating
its rate of dissociation from the channel.
patterns of impulses and membrane depolarizations caused by abnormal expression of
Na+
channels rather than from selective sensitivity of certain subtypes
of channels ( Fig. 14-10
).