|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Multiple sclerosis (MS), the most common demyelinating disease of the CNS, affects about 1 million people worldwide. The cause of MS is not known. Infectious agents, genetic predisposition, autoimmune reactions to antigens, and channel disease have been implicated in the etiology of MS.[825] [826] [827] [828] [829] [830] Recently, it has been hypothesized that MS is a sexually transmitted disease.[828] Paralysis, sensory disturbances, autonomic disturbances, lack of coordination, and visual impairment are common features.[831] Axon loss correlates with permanent functional deficit.[832] It has been demonstrated that the CSF of patients with MS may contain a sodium channel-blocking factor (a local anesthetic-like factor).[833] [834] [835] [836] This factor could explain the paresis seen in this disorder.[836] Therefore, it appears that channelopathies play an important role in the pathogenesis of this disorder.[827] In MS, the characteristics of individual skeletal muscles are similar to those observed in disuse myopathy. [837]
Autonomic dysfunction is seen in a significant number of patients with MS.[838] [839] During anesthesia, careful attention should be paid to maintenance of adequate preload, temperature control, postural changes, blood loss, and peak airway pressure during mechanical ventilation. Patients with autonomic dysfunction demonstrate an exaggerated response to α-sympathomimetics.[840]
No conclusive evidence has shown that the stress of surgery and anesthesia may increase the rate of relapse in patients with MS.[841] [842] The use of regional anesthesia in MS is more controversial. Both lumbar epidural anesthesia and subarachnoid anesthesia have safely been used in patients with MS.[843] However, some evidence suggests that hyperthermia[842] or higher concentrations of local anesthetic[841] may increase the relapse rate. In one study in which patients received either 0.5% or 0.25% bupivacaine for epidural anesthesia, relapses occurred only in patients receiving the higher dose of local anesthetic.[841]
As with any patient with denervation or disuse, or both, there may be upregulation in nAChR numbers and increased sensitivity to depolarizing neuromuscular blockers.[17] In this case, the patient is at risk for hyperkalemia after the administration of succinylcholine. Paradoxical reports have described increased sensitivity to nondepolarizing neuromuscular blockers in patients with MS, probably because of reduced muscle mass or reduced margin of safety for neuromuscular transmission.[844] Denervation is known to induce a reduction in the resting potential, and this decreased resting potential will significantly contribute to muscle weakness.[17] [845] [846] In these patients, small doses of short-acting neuromuscular
The motor neuron diseases are a group of diverse disorders characterized by muscle weakness, atrophy, spastic paralysis, or a combination of these findings as a result of involvement of lower or upper motor neurons.[17] Amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig's disease, is the most common motor neuron disease and has an incidence of 2 to 4 in 100,000. ALS is a progressive disease characterized by degeneration of cortical, brainstem, and spinal motor neurons.[847] The cognitive and sensory systems are usually spared. Kennedy's disease (spinobulbar muscular atrophy) affects lower motor neurons only. In contrast, hereditary spastic paraplegia involves upper motor neurons.[17]
Several mechanisms have been proposed to account for the progressive motor neuron death evident in ALS, including oxidative stress,[848] neurofilament damage,[849] mitochondrial abnormalities, [850] glutamate-mediated excitotoxicity,[851] and altered responses to hypoxia.[852] A role of oxidative stress has been suggested because mutations in the gene for Cu2+ /Zn2+ superoxide dismutase (SOD1), which catalyzes conversion of the O2 • radical to O2 and H2 O2 , have been identified in some 3% of all ALS cases.[848] In addition, antibodies to voltage-gated Ca2+ channels have been identified in ALS patients.[853] Treatment of ALS is aimed at symptomatic support.[847] Stem cell therapy may offer hope in the future.[854]
Patients with motor neuron disease are at risk for hyperkalemia after the administration of succinylcholine because of upregulation of nAChRs.[855] ALS patients have, in addition, presynaptic impairment of neuromuscular transmission, [856] which explains their hypersensitivity to nondepolarizing neuromuscular blockers.[857]
Respiratory muscle weakness frequently develops in patients with ALS, and most die of pulmonary complications.[858] [859] Particularly in late stages of the disease, patients may be cachectic from inadequate nutrition and demonstrate reduced plasma protein binding for many of the anesthetic drugs.[17] These patients have reduced respiratory muscle reserve and abnormal airway protective reflexes and are at increased risk for respiratory depression and aspiration secondary to the use of sedative and anesthetic drugs.[17] Epidural anesthesia has been used in ALS patients without reported untoward effects. [860] [861]
ALS is not believed to be associated with significant autonomic dysfunction. There is, however, evidence of sympathetic hyperactivity[862] and autonomic failure[863] accompanied by reduced baroreflex sensitivity.[864]
Guillain-Barré syndrome (GBS) is now considered to be a collection of diverse disorders with several clinical manifestations[865] [866] and not simply as it was first described—"syndrome of symmetric, rapidly evolving flaccid paralysis and areflexia."[866] In addition to the demyelination seen in GBS, channelopathies have been identified. [827] GBS is not uncommon and has an incidence of 4 in 10,000.[865] Deaths are usually related to respiratory or autonomic dysfunction.
A sodium channel-blocking factor (a local anesthetic-like factor) has been identified in the CSF of patients with GBS.[833] [834] [836] This factor could contribute to the paralysis seen in this disorder.[836] There is also strong evidence for an association between certain infections and GBS.[865] [867] Bacterial antigens are capable of initiating an immune response that targets similar moieties on nerve fibers[866] or blocks both presynaptic voltage-gated calcium channels[868] and postsynaptic nAChR channels,[869] thereby leading to neuromuscular weakness. GBS patients commonly have symptomatic improvement after plasmapheresis. [870]
Demyelination or axonal degeneration produces functional denervation of the muscle and upregulation of nAChRs at the postsynaptic membrane. Succinylcholine is contraindicated because of the risk of hyperkalemic cardiac arrest.[871] [872] This risk may persist after recovering from the symptomatic neurologic deficit.[873] Increased sensitivity to nondepolarizing neuromuscular blockers is expected in these patients because of the loss of motor units and channels blockade at the neuromuscular junction. [869] [872] [874]
Autonomic dysfunction is seen in approximately 60% of patients. [875] Asystole was reported after eyeball pressure, carotid sinus massage, and tracheal suction in patients with GBS.[876] During anesthesia, careful attention should be paid to maintenance of adequate preload, temperature control, postural changes, blood loss, and peak airway pressure during mechanical ventilation. Patients with autonomic dysfunction demonstrate an exaggerated response to α-sympathomimetics.[840]
Regional anesthesia is not contraindicated, although patients with GBS are sensitive to local anesthetics,[872] [877] probably secondary to the presence of the sodium channel-blocking factor.[833] [834] [836] Because of the high incidence of autonomic instability, the slower onset of an epidural block may be preferable to the rapid onset of subarachnoid anesthesia.[17] GBS has been reported in four patients 1 to 2 weeks after epidural anesthesia. It was suggested that local trauma to nerve roots may initiate a cascade of immunologic events that result in demyelinating neuropathy in these patients.[878]
Charcot-Marie-Tooth disease (CMTD; hereditary motor and sensory demyelinating polyneuropathy) is the most frequently occurring inherited peripheral neuropathy, with an incidence of 1 in 2500.[879] It has diverse genetic (autosomal dominant, X-linked, or autosomal recessive) and clinical manifestations.[880] [881] Three genes responsible for CMTD type 1 have been identified: peripheral myelin protein 22 and myelin protein zero for the autosomal dominant form and connexin 32 for the X-linked dominant variant.[882] The latter variant encodes a gap junction protein.[881] Gap junctions are aggregations of channels made of proteins called connexins that are present in the plasma membrane.[883] In the nervous system, gap junctional channels play an essential role in the propagation of action potentials and in allowing rapid exchange of ions and nutrients.[884] Failure of gap junctions leads to impaired Schwann cell function and demyelination.
Peroneal nerve atrophy leading to weakness in the anterior and lateral compartments is the most common clinical manifestation in CMTD, but considerable variability exists in the pattern of atrophy. The sensory disturbance is milder than the motor disturbance.[17] Autonomic disturbances are occasionally reported.[885] Pregnancy may be associated with exacerbations of CMTD, probably because of hormonal changes.[886] [887] Respiratory insufficiency has also been described in patients with CMTD.[888]
CMTD patients have no evidence of a prolonged response to nondepolarizing neuromuscular blockers.[889] [890] Although drugs that trigger malignant hyperthermia have been used in CMTD patients without untoward effects,[891] [892] episodes of malignant hyperthermia have been reported in these patients.[893] [894] Therefore, it is advisable to avoid using drugs known to trigger malignant hyperthermia.[17]
Patients with CMTD have been reported to be sensitive to the effects of thiopental.[885] However, propofol infusion has been safely used in these patients.[889] [895] Respiratory insufficiency, vocal cord paresis, and cardiac conduction abnormalities have also been described in patients with CMTD.[888] [896] [897] [898] [899] [900] Epidural anesthesia has been used successfully for labor and delivery in patients with CMTD. [901] [902]
Muscular dystrophies are a diverse group of genetically determined disorders of skeletal muscle and in some cases cardiac muscle.[903] They are characterized by muscle fiber necrosis and progressive muscle weakness. [903] The current classification of these disorders relies on molecular, genetic, and protein biochemical characterization ( Table 13-21 ).[903]
In humans, mutations in the gene encoding the dystrophin-glycoprotein
complex cause muscular dystrophy.[915]
This complex
is essential in maintaining the functional integrity of sarcolemma,[915]
[916]
and dystrophic muscles are susceptible to
mechanical
injury as manifested by repeated necrosis and regeneration of muscle fibers.[917]
Loss
| Diseases | Mode of Inheritance | Molecular Etiology | Reference |
|---|---|---|---|
| Duchenne | X-linked recessive | Absence of dystrophin | [904] [924] |
| Becker | X-linked recessive | Reduced dystrophin | [905] [924] |
| Emery-Dreifuss | X-linked recessive | Emerin | [906] |
|
|
Autosomal dominant | Lamin A/C | [907] |
| Limb-girdle | Autosomal dominant or recessive | Sarcoglycan deficiency | [908] |
| Congenital muscular dystrophy (CMD) | Autosomal recessive |
|
|
| Classic CMD |
|
Laminin α2 chain | [909] |
| Fukuyama CMD |
|
Fukutin | [910] |
| α7 Integrin congenital myopathy |
|
α7 Integrin (laminin receptor) | [911] |
| Facioscapulohumeral | Autosomal dominant | 4q35 rearrangements | [912] |
| Myotonic dystrophy (MD) | Autosomal dominant |
|
|
| MD1 |
|
19q13 rearrangements | [913] |
| MD2 |
|
3q21 rearrangements | [914] |
| Modified from Naguib M, Flood P, McArdle JJ, et al: Advances in neurobiology of the neuromuscular junction: Implications for the anesthesiologist. Anesthesiology 96:202–231, 2002. | |||
Duchenne's muscular dystrophy is one of the most common genetic diseases in humans, with an incidence of 1 in 3500 male births.[915] It is characterized by progressive proximal weakness beginning in early childhood and progressive cardiomyopathy.[922] Cognitive impairment is also observed and is probably caused by a lack of dystrophin in the neuronal membrane.[923] Death occurs before 30 years of age as result of respiratory or cardiac failure.[922]
Becker's muscular dystrophy is milder and affects 1 in 30,000 male births.[922] Both Duchenne's and Becker's muscular dystrophies are due to an X-linked recessive mutation in the dystrophin gene.[924] In Duchenne's muscular dystrophy, dystrophin is usually absent, whereas in Becker's muscular dystrophy, the protein is present but qualitatively and quantitatively abnormal.[904] [925] Onset in childhood may occur as late as 16 years. Cardiomyopathy is present in approximately 15% of patients younger than 16 years and in 75% of those older than 40 years.[922]
Limb-girdle dystrophy is similar to Duchenne's dystrophy and is found most commonly in families in North Africa. Limb-girdle muscular dystrophy may result from autosomal dominant (type 1) or autosomal recessive (type 2) mutations and affects approximately 1 in 20,000 people.[908] [915]
Congenital muscular dystrophy has the worst prognosis. Affected infants at birth have hypotonia, weakness, and respiratory and swallowing abnormalities. [909] Fukuyama-type congenital muscular dystrophy, one of the most common (0.7 to 1.2 per 10,000 births) autosomal recessive disorders in Japan, is associated with severe mental retardation and cortical dysgenesis.[926] This syndrome
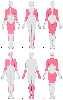 Figure 13-35
Distribution of predominant muscle weakness in different
types of dystrophy. A, Duchenne type and Becker type;
B, Emery-Dreifuss; C,
limb girdle; D, facioscapulohumeral; E,
distal; and F, oculopharyngeal. (Redrawn
from Emergy AE: The muscular dystrophies. BMJ 317:991–995, 1998.)
Figure 13-35
Distribution of predominant muscle weakness in different
types of dystrophy. A, Duchenne type and Becker type;
B, Emery-Dreifuss; C,
limb girdle; D, facioscapulohumeral; E,
distal; and F, oculopharyngeal. (Redrawn
from Emergy AE: The muscular dystrophies. BMJ 317:991–995, 1998.)
Recently, it has been possible to rescue dystrophic symptoms in a mouse model of congenital muscular dystrophy by muscle-specific overexpression of an agrin minigene that replaces the missing link between the basement membrane and the muscle fiber.[928] In addition, intravenous injection of stem cells into a mouse model of congenital muscular dystrophy resulted in partial restoration of dystrophin expression in the affected muscle.[929] Therefore, these novel approaches may provide new therapeutic tools to restore muscle function in human muscular dystrophies.
Emergy-Dreifuss muscular dystrophy is characterized by early contractures of the elbows and Achilles tendons and wasting in the humeroperoneal muscles; it has an incidence of 1 in 33,000 male births. Cardiomyopathy and conduction blocks are common and can be life-threatening.[930] Two modes of inheritance exist, X-linked[906] and autosomal dominant.[907] Both forms of the disease are clinically identical.
Facioscapulohumeral muscular dystrophy is a rare variant of muscular dystrophy with an incidence of 10 to 20 cases per million.[931] It is usually manifested in late childhood as facial and scapulohumeral weakness, but cardiac involvement is generally absent. Retinal vasculopathy and sensorineural hearing loss may develop.[912] [922] Oculopharyngeal dystrophy is another rare variant of muscular dystrophy that is characterized by progressive dysphagia and ptosis.
Myotonic dystrophy is the most common form of muscular dystrophy in adults, with an incidence of 1 in 8000. It can result from a mutation on either chromosome 19q13 (myotonic dystrophy type 1)[913] or chromosome 3q21 (myotonic dystrophy type 2).[914] Myotonic dystrophy type 1 is caused by a CTG expansion in the 3' untranslated region of the dystrophia myotonica-protein kinase gene (DMPK).[913] [932] DMPK may be involved in cellular Ca2+ homeostasis.[933] Abnormalities in sarcoplasmic reticulum Ca2+ transport have been noted in muscle fibers with myotonic dystrophy.[934]
Myotonic dystrophy is a dominantly inherited disease characterized by myotonia, progressive myopathy, insulin resistance, defects in cardiac conduction, neuropsychiatric impairment, cataracts, testicular atrophy, and frontal balding in males.[935] Patients with myotonic dystrophy have increased mortality from respiratory complications secondary to aspiration as a result of their muscle weakness, as well as cardiac dysrhythmias. The severity of the symptoms is somewhat related to the number of trinucleotide repeats in DMPK.[936]
In dystrophic muscle, postsynaptic nAChRs are expressed as a mixture of fetal- and mature-type receptors.[17] [937] Expression of fetal nAChR is not a characteristic of dystrophy but a consequence of muscle regeneration.[937] Succinylcholine is contraindicated in these patients because of the risk of hyperkalemic cardiac arrest and rhabdomyolysis.[658] [659] This response has led to a Food and Drug Administration-mandated warning against the use of succinylcholine in pediatric patients because of potential mortality in those with clinically inapparent muscular dystrophies. In addition, the incidence of malignant hyperthermia is increased in patients with muscular dystrophies.[938] [939]
Resistance to nondepolarizing neuromuscular blockers would be expected on the basis of the reduced sensitivity of fetal nAChRs to competitive antagonists. [17] However, clinically the reverse is seen[940] [941] [942] and has been attributed to the underlying muscle wasting and reduced ability to produce contractile force.[943] Other reports, however, indicate a normal response.[944] Buzello and coauthors[945] reported that the response of patients with myotonic dystrophy to neostigmine is unpredictable.[945] Attempts to reverse residual nondepolarizing blockade in one patient with 1.0 mg neostigmine were only partially effective, and the administration of a second dose (0.5 mg) produced long-lasting muscle weakness.[945] A tonic response to neostigmine was noted in another patient with myotonic dystrophy.[945] It is advisable to avoid using anticholinesterases in these patients.
Rhabdomyolysis, with or without cardiac arrest, can occur with inhaled anesthetics, even if succinylcholine is avoided.[946] [947] [948] This complication raises major concern regarding the safety of inhaled anesthetics in muscular dystrophy patients.[949] [950]
Mathieu and colleagues[951] identified other potential anesthetic problems in patients with myotonic dystrophies, such as glossal hypertrophy, delayed gastric emptying, contractures, spinal deformities, impaired respiratory function, and cardiomyopathy. Therefore, preoperative pulmonary and cardiac status should be evaluated carefully in these patients. The need for a cardiac pacemaker should also be assessed preoperatively in patients with high-degree atrioventricular blocks. Weak ventilatory muscles, velopalatal insufficiency, and delayed gastric emptying are expected to increase the risk of aspiration and pneumonia. [858] [952] [953] [954] Clinical deterioration may occur in pregnancy, probably because of hormonal changes.[955] Insulin resistance syndrome is also noted in patients with myotonic dystrophy[956] and is probably the result of a lack of insulin receptors in the muscle fiber membrane. [957]
Case reports have described increased sensitivity to thiopental and propofol in myotonic dystrophy patients.[958] [959] [960] However, further studies have not confirmed these reports.[950] [951] [961] In a recent report, neither exaggerated reactions nor hemodynamic instability was observed in 13 patients with myotonic dystrophy anesthetized by continuous propofol infusion, fentanyl, atracurium, and nitrous oxide.[950] The exaggerated physiologic responses to intravenous anesthetics may be related to the severity of the disease.
Regional anesthesia and total intravenous anesthesia with propofol, opioid anesthesia, and a nondepolarizing neuromuscular blocker appear to be safe and effective anesthetic techniques in patients with muscular dystrophy.[942] [950] [962] [963] [964] It is advisable to use short-acting nondepolarizing neuromuscular blockers and monitor neuromuscular function until full recovery to avoid the administration of anticholinesterases in these patients.
Blood loss in patients with Duchenne's muscular dystrophy is significantly greater than in those with spinal muscular atrophy undergoing scoliosis surgery. [965] The difference in blood loss is probably related to an inadequate vasoconstrictive response because of a lack of dystrophin, a reduction in neuronal nitric oxide synthase, and platelet dysfunction.[919] [966]
Mitochondrial myopathies are a clinically and biochemically heterogeneous group of disorders with genetic abnormalities that involve either a mitochondrial or a nuclear gene[967] ; the prevalence is 7 per 100,000.[968] This disorder targets metabolically active organs such as the liver, the brain, and skeletal muscles because they contain the largest number of mitochondria.[969] [970]
Mitochondrial myopathies are often associated with abnormal proliferation of mitochondria, which accumulate beneath the sarcolemma and between muscle fibers. Histologically, staining affected muscles with Gomori modified trichrome gives the characteristic ragged red fibers. However, ragged red fibers are not pathognomonic of a mitochondrial DNA mutation.[971]
Both isolated myopathies and several multisystem syndromes have been identified. The syndromes, which are defined by characteristic clinical manifestations in addition to mitochondrial myopathy, are chronic progressive external ophthalmoplegia, including Kearns-Sayre syndrome, MELAS syndrome (mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes), MERRF syndrome (myoclonus epilepsy and ragged red fibers), MNGIE syndrome (myopathy, external ophthalmoplegia, neuropathy, and gastrointestinal encephalopathy), and NARP syndrome (neuropathy, ataxia, and retinitis pigmentosa). Acquired mitochondrial myopathy has been associated with the use of zidovudine, an antiretroviral drug that depletes muscle mitochondrial DNA.[972]
Patients with mitochondrial disease may have lactic acidosis in the absence of hypoxia or sepsis.[973] Acid-base status should be monitored intraoperatively. Catecholamines, theophylline, nitroprusside, and prolonged infusion of propofol have been reported[974] [975] [976] to increase lactate concentrations by inducing transient abnormalities in oxidative phosphorylation. [973] Metabolic acidosis is also noted after exercise, fasting, and short-term use of a new formulation of propofol.[977] The incidence of diabetes mellitus is relatively high in patients with mitochondrial diseases.[978]
Respiratory failure may result from an impaired ventilatory response to hypercapnia and hypoxia,[979] muscle weakness, [980] diaphragmatic paralysis, or any combination of these conditions.[973] Sleep apnea is not uncommon in patients with mitochondrial disease.[981] Hypertrophic cardiomyopathies, cardiac conduction defects, and hypertension are also seen in mitochondrial disorders.[982] [983] Bulbar muscle involvement is likely to increase the risk of aspiration in patients with mitochondrial disorders.[984]
Although it has been suggested that mitochondrial myopathy does not involve the neuromuscular junction,[985] increased sensitivity to different nondepolarizing neuromuscular blockers has been demonstrated in patients with mitochondrial myopathies.[986] [987] This enhanced sensitivity is of a magnitude similar to that observed in myasthenia gravis.[986] Increased sensitivity to succinylcholine was also noted in these patients.[988] The association between malignant hyperthermia and mitochondrial myopathies is not clear, but published reports indicate a possible association.[989] [990] [991] Intrathecal and epidural anesthesia appears to be safe in patients with mitochondrial myopathies. [992] [993]
Cell membranes are composed of two lipid layers, which are not permeable to ions. However, cell membranes have channels that allow ions to diffuse in order to generate an action potential.[994] Siegelbaum and Koester[995] recognized the functions of ion channels as follows: "(1) they conduct ions; (2) they recognize and select among specific ions; and (3) they open and close in response to specific electrical, mechanical or chemical signals." In the neuromuscular junction, different channels are present at the prejunctional and postjunctional sites. These channels play an integral role in maintaining the functional integrity of the neuromuscular junction in health. Disorders of channel function
Waxman[998] pointed out that there are three main causes of channelopathy and proposed the following classification: (1) acquired (immune mediated and toxic), (2) genetic (caused by mutations in ion channel genes[994] ), and (3) transcriptional channelopathies. [998] In immune-mediated and toxic channelopathies, binding of antibodies or toxins to channels alters their function.[866] Examples of acquired (immune mediated) channelopathies affecting the neuromuscular junction are myasthenia gravis (MG), Lambert-Eaton myasthenic syndrome (LEMS), and Isaacs' syndrome or neuromyotonia. A list of hereditary channelopathies that involve the neuromuscular junction is provided in Table 13-22 . Waxman[998] pointed out that transcriptional channelopathies are due to "dysregulated expression of non-muted genes." Changes in Na+ channel transcription have recently been implicated in MS.[999]
For a more extensive account of this subject, the reader is referred to reviews by Hoffman,[1010] Cooper and Jan,[1011] Lehmann-Horn and Jurkat-Rott,[1012] and Kleopa and Barchi.[1013] Based on the current evidence, [1010] [1011] [1012] [1014] the role of ion channels in neuromuscular transmission can be summarized as follows:
| Disease | Ion Channel Subunit | Gene | Reference |
|---|---|---|---|
| Myotonia congenita (dominant and recessive) | Voltage-gated Cl- channel | CLCN1 | [1000] |
|
|
|
|
[1001] |
| Hyperkalemic periodic paralysis | Voltage-gated Na+ channel | SCN4A | [1002] |
| Paramyotonia congenita |
|
|
[1003] |
| Potassium-aggravated myotonia |
|
|
[1004] |
| Hypokalemic periodic paralysis type 1 | Voltage-gated Ca2+ channel (dihydropyridine receptor) | CACNA1S | [1005] |
| Hypokalemic periodic paralysis type 2 | Voltage-gated Na+ channel | SCN4A | [1006] |
| Malignant hyperthermia | Ligand-gated Ca2+ channel | RYR1 | [1007] |
| Central core disease |
|
|
[1008] |
| Congenital myasthenic syndromes | nAChR channel | CHRNA1 | [1009] |
| X-linked Charcot-Marie-Tooth disease | Connexin | GJB1 (Cx32) | [882] |
| nAChR, nicotinic acetylcholine receptor. | |||
| Modified from Naguib M, Flood P, McArdle JJ, et al: Advances in neurobiology of the neuromuscular junction: Implications for the anesthesiologist. Anesthesiology 96:202–231, 2002. | |||
The large number (10,000/µm2 ) of nAChRs in the postsynaptic muscle membrane is crucial for maintaining normal neuromuscular function and for allowing a margin of safety in neuromuscular transmission.[1018] Although it is now established that MG is due to autoantibodies to
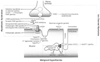 Figure 13-36
Disorders of channel function (channelopathies) that
cause myasthenic syndromes, myotonias, malignant hyperthermia (MH), and central core
disease (CCD). AChE, acetylcholinesterase; DHP receptor, dihydropyridine receptor;
MuSk, muscle-specific kinase; nAChR, nicotinic acetylcholine receptor; RyR, ryanodine
receptor; SV, synaptic vesicle; VGCC (P/Q), voltage-gated calcium channel (P/Q type);
VGCLC, voltage-gated chloride channel; VGKC, voltage-gated potassium channel; VGNC,
voltage-gated sodium channel.
Figure 13-36
Disorders of channel function (channelopathies) that
cause myasthenic syndromes, myotonias, malignant hyperthermia (MH), and central core
disease (CCD). AChE, acetylcholinesterase; DHP receptor, dihydropyridine receptor;
MuSk, muscle-specific kinase; nAChR, nicotinic acetylcholine receptor; RyR, ryanodine
receptor; SV, synaptic vesicle; VGCC (P/Q), voltage-gated calcium channel (P/Q type);
VGCLC, voltage-gated chloride channel; VGKC, voltage-gated potassium channel; VGNC,
voltage-gated sodium channel.
MG is an antibody-mediated autoimmune disease targeted against the α-subunit of nAChRs at the neuromuscular junction; it has a prevalence of 0.25 to 2.00 per 100,000 people.[1018] [1019] [1020] [1021] In MG, the number of functional nAChRs is markedly decreased as a result of (1) cross-linking of antibodies to the receptors[1022] and (2) focal membrane lysis caused by complement fixation.[1021] [1023] [1024] The condition results in muscular weakness and fatigability.[1021]
Antibodies to the nAChR are present in about 80% of patients with MG.[1025] [1026] In the remaining 20% of patients (called seronegative patients), nAChR antibodies are not detectable.[1027] Recently, another form of antibodies has been identified in seronegative MG patients. In about 70% of seronegative (but not seropositive) MG patients, the muscle-specific receptor tyrosine kinase (MuSK) has been identified as the target for autoantibodies (see Fig. 13-36 ).[1028] MuSK mediates the agrin-induced clustering of AChRs during synapse formation and is also expressed at the mature neuromuscular junction.[17] [1029] Interestingly, antibodies from MG patients do not cross-react with the α3 -subunit of the nAChR that is found principally in the autonomic nervous system or the α4 β2 nAChRs that occur in the CNS. Perhaps this explains the lack of autonomic and CNS symptoms in typical MG.[1030]
The triggers for the immune response in MG are largely unknown. The thymus has been implicated because approximately 70% of MG patients have thymic lymphoid follicular hyperplasia with germinal centers that produce antibodies to nAChRs.[1020] In a small percentage of MG patients, autoantibodies develop as part of a paraneoplastic syndrome.[1020] About 12% of patients with MG have a thymoma, whereas 30% to 50% of patients with a thymoma suffer from MG.[1020] It is believed that antibodies to nAChRs are produced in other locations because thymectomy does not cure MG and does not protect against the occurrence of MG.[1020] [1031] Fetal-type nAChRs,
| Syndrome | Location | Mechanism | Etiology |
|---|---|---|---|
| Lambert-Eaton myasthenic syndrome | Presynaptic | Autoimmune | Antibodies to voltage-gated calcium channels at the motor nerve terminal |
| Congenital myasthenic syndromes |
|
Genetic |
|
| Choline acetyltransferase deficiency | Presynaptic |
|
Mutations in choline acetyltransferase |
| Acetylcholinesterase deficiency | Synaptic |
|
Mutations in the gene encoding the collagenic tail subunit (ColQ) of the enzyme that anchors acetylcholinesterase in the synaptic cleft |
| Slow- and fast-channel syndromes | Postsynaptic |
|
Mutations in nAChR genes |
| nAChR deficiency | Postsynaptic |
|
Mutations in nAChR genes or in rapsyn |
| Myasthenia gravis | Postsynaptic | Autoimmune |
|
| Seropositive |
|
|
Antibodies to nAChRs |
| Seronegative |
|
|
Antibodies to MuSK |
| MuSK, muscle-specific kinase; nAChR, nicotinic acetylcholine receptor. | |||
Electron microscopic studies show that the postsynaptic membrane has a simplified appearance with little folds[1033] associated with a marked reduction in the clustering of nAChRs to approximately 30% of those seen in normal neuromuscular junctions.[1040] Although acetylcholine sensitivity is reduced, a compensatory increase in the release of acetylcholine occurs at the neuromuscular junction in both experimental models of MG[1034] and muscle biopsy specimens from patients with MG.[1035]
Improvement in strength after the intravenous injection of edrophonium (Tensilon) helps confirm the diagnosis of MG. After a test dose of 1 to 2 mg, a total dose of 10 mg is administered intravenously. A positive response is expected within 5 minutes. No specific immunotherapy is available for MG. Nonspecific immunosuppression with steroids and other drugs and plasmapheresis are often combined with thymectomy and symptomatic treatment with anticholinesterases.
LEMS is an example of an acquired (immune mediated) channelopathy that results from autoantibodies targeting the presynaptic voltage-gated Ca2+ channels and possibly another presynaptic component (such as synaptotagmin), and as a consequence, acetylcholine release is reduced.[1026] [1036] [1037] [1038] Synaptotagmin plays an important role in synaptic vesicle fusion and fast release of acetylcholine.[17] [1039] Approximately 60% of LEMS patients show a paraneoplastic response, often in association with small cell carcinoma of the lung.[1026] LEMS is also characterized by weakness and fatigability.
Although both LEMS and MG are autoimmune diseases, they have several differences: (1) in LEMS, the presynaptic site of the neuromuscular junction is the target for autoantibodies, whereas postsynaptic nAChRs are the target in MG; (2) autonomic disturbances are seen in about 30% of patients with LEMS but not in patients with MG; (3) unlike MG, anticholinesterases are of little therapeutic value in LEMS[1036] ; (4) improvement in muscle strength is seen after exercise in LEMS as a result of summation of presynaptic Ca2+ signals and improved acetylcholine release,[1040] but in MG improvement occurs after rest; (5) LEMS is differentiated from MG by electromyography, in which facilitation of the electromyographic response, rather than fade, occurs during high-frequency (30 to 50 Hz) stimulation; and (6) the two diseases can also be differentiated by antibody titer to specific channels. The acetylcholine contents and the architecture of the neuromuscular junction are normal in diseased nerve endings in LEMS.
Plasmapheresis or intravenous immunoglobulin can often give transient improvement in LEMS.[1041] Treatment with 3,4-diaminopyridine results in significant improvement in symptoms in patients with LEMS.[1042] Pyridostigmine potentiates the response to 3,4-diaminopyridine in many patients. [1042] 3,4-Diaminopyridine blocks potassium channels and thereby prevents potassium efflux. Prevention of potassium efflux increases the action potential duration, which in turn prolongs the activation of voltage-gated Ca2+ channels, along with a concomitant increase in intracellular Ca2+ concentration and acetylcholine release.
Congenital myasthenic syndromes (CMSs) are diverse disorders characterized by muscle weaknesses and fatigability (like MG) and caused by congenital defects in different components of the neuromuscular junction (see Fig. 13-36 ).[994] [1043] [1044] [1045] Inherited mutations are seen in the presynaptic (synaptic vesicles, choline acetyltransferase), synaptic (acetylcholinesterase), or postsynaptic
MG and CMSs have several differences: (1) unlike MG, antibodies against the nAChRs are not present and immunosuppressive therapy is not effective in CMSs, and (2) in contrast to neonatal MG, which is caused by passive transfer of anti-nAChR antibodies from a myasthenic mother to the fetus, the mother of a CMS patient does not have myasthenia.
An increase in the affinity of the nAChR for acetylcholine is seen in SCCMSs.[1009] The net effect of such gain-of-function mutations is to prolong the open state of the nAChR.[17] Such prolongation allows what is normally physiologic activation of the neuromuscular junction to overload the postsynaptic region with Ca2+ and initiates necrosis. [17] Activation of nitric oxide synthase at the neuromuscular junction can also contribute to free radical damage of the end plate. Nitric oxide synthase inhibitors may be of value in these patients.[1050] Patients with SCCMSs are significantly improved by quinidine sulfate because it normalizes the open duration of slow-channel mutants.[1051]
Loss-of-function mutations, seen in the α- and epsilon-subunits, decrease the rate of channel opening and increase the closure rate.[1052] [1053] This loss of nAChR function reduces the safety factor for synaptic transmission.[17]
For detailed reviews, see Baraka[1054] and Abel and Eisenkraft.[1055] Preoperative assessment and preparation of an MG patient should include (1) consultation with the patient's neurologist to learn of the recent history and progress of management; (2) preoperative drug therapy (such as pyridostigmine and immunosuppression drugs) and the potential impact of this drug therapy on responses to neuromuscular blockers; (3) counseling and preparation of the patient for possible postoperative endotracheal intubation and mechanical ventilation; and (4) optimization of medical management for myasthenia, which may include preoperative plasmapheresis and continuation of the anticholinesterase therapy. Pyridostigmine therapy should be continued preoperatively.
Patients with bulbar involvement are at increased risk for respiratory depression and aspiration, especially during myasthenic crises.[1056] It has been reported that 25% of myasthenic crisis episodes were associated with radiographic evidence of aspiration pneumonia.[1057] About 33% of the patients in crisis had severe oropharyngeal weakness.[1057] Pulmonary function tests, including flow-volume loops, may be necessary to predict the need for mechanical ventilation postoperatively.[1058]
Because of the decreased number of nAChRs, myasthenic patients are resistant to succinylcholine ( Fig. 13-37 ).[1059] On the other hand, butyrylcholinesterase activity may be
 Figure 13-37
Succinylcholine dose-response curves in normal and myasthenic
patients. (Redrawn from Eisenkraft JB, Book WJ, Mann SM, et al: Resistance
to succinylcholine in myasthenia gravis: A dose-response study. Anesthesiology
69:760–763, 1988.)
Figure 13-37
Succinylcholine dose-response curves in normal and myasthenic
patients. (Redrawn from Eisenkraft JB, Book WJ, Mann SM, et al: Resistance
to succinylcholine in myasthenia gravis: A dose-response study. Anesthesiology
69:760–763, 1988.)
The loss of approximately 70% of the postsynaptic nAChRs means that myasthenic patients have a marked reduction or even total loss of the safety margin for neuromuscular transmission. Therefore, it is not unexpected that patients with MG are extremely sensitive to nondepolarizing neuromuscular blockers ( Fig. 13-39 and Fig. 13-40 ). [1054] [1063] [1064] [1065] The effective dose of vecuronium is 250% greater in control patients than in MG patients,[1066] but this does not mean that nondepolarizing neuromuscular blockers are contraindicated in these patients. With careful titration and adequate monitoring of neuromuscular function, nondepolarizing agents have been used safely in myasthenic patients undergoing thymectomy.[1054] [1058] [1063] [1064] Long-acting neuromuscular blocking drugs should be avoided in these patients. Intermediate-acting drugs should be used in low dosage as guided by monitoring with a nerve stimulator. About one tenth to one fifth the ED95 should be given as a test dose to estimate the patient's requirement. Individual response will vary from extreme sensitivity, such that the test dose is all that is needed, to nearly normal relaxant requirements.
Pyridostigmine will modify the response to relaxants as follows: (1) the sensitivity to nondepolarizers will be diminished, (2) the response to succinylcholine or mivacurium may be prolonged, and (3) reversal of residual block at the end of the procedure may be ineffective because much acetylcholinesterase inhibition already
 Figure 13-38
Electromyographic response to ulnar nerve stimulation
by train-of-four stimulation every 20 seconds. Shown is the effect of 1.5 mg/kg
succinylcholine in three myasthenic patients with differing butyrylcholinesterase
activity. Upper tracing, butyrylcholinesterase =
5.16 U/mL; middle tracing, butyrylcholinesterase
= 1.45 U/mL; lower tracing, butyrylcholinesterase
= 0.73 U/mL. (From Baraka A: Suxamethonium block in the myasthenic patient.
Correlation with plasma cholinesterase. Anaesthesia 47:217–219, 1992.)
Figure 13-38
Electromyographic response to ulnar nerve stimulation
by train-of-four stimulation every 20 seconds. Shown is the effect of 1.5 mg/kg
succinylcholine in three myasthenic patients with differing butyrylcholinesterase
activity. Upper tracing, butyrylcholinesterase =
5.16 U/mL; middle tracing, butyrylcholinesterase
= 1.45 U/mL; lower tracing, butyrylcholinesterase
= 0.73 U/mL. (From Baraka A: Suxamethonium block in the myasthenic patient.
Correlation with plasma cholinesterase. Anaesthesia 47:217–219, 1992.)
Different anesthetic techniques have been used in myasthenic patients. Although surgical relaxation can be provided for a myasthenic patient with only a potent inhaled anesthetic without neuromuscular blockers, this technique may be associated with slow recovery from anesthesia. In addition, myasthenic patients are more sensitive than normal to the neuromuscular depressant effects of halothane and isoflurane.[1068] [1069] [1070] Therefore, it may be safer to intubate the trachea and provide surgical relaxation with the aid of nondepolarizing neuromuscular blockers in these patients than to use deep inhalation anesthesia.
A thoracic epidural anesthetic in combination with balanced general anesthesia provides excellent analgesia both intraoperatively and during the period after
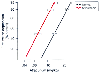 Figure 13-39
Cumulative dose response for atracurium in patients with
myasthenia gravis. (Redrawn from Smith CE, Donati F, Bevan DR: Cumulative
dose-response curves for atracurium in patients with myasthenia gravis. Can J Anaesth
36:402–406, 1989.)
Figure 13-39
Cumulative dose response for atracurium in patients with
myasthenia gravis. (Redrawn from Smith CE, Donati F, Bevan DR: Cumulative
dose-response curves for atracurium in patients with myasthenia gravis. Can J Anaesth
36:402–406, 1989.)
Patients with LEMS are sensitive to both depolarizing and nondepolarizing neuromuscular blockers.[1074] In fact, patients with LEMS have significantly greater sensitivity to nondepolarizing neuromuscular blockers than those with MG do.[1075] In patients with LEMS, neostigmine is ineffective as an antagonist for residual neuromuscular block.[1076] It has been suggested that a combination of an anticholinesterase and 4-aminopyridine might be of value in
 Figure 13-40
Electromyographic response to ulnar nerve stimulation.
Injection of 0.1 mg/kg vecuronium in a normal patient resulted in a slow onset of
nondepolarizing block (upper trace), whereas injecting
one tenth the dose (0.01 mg/kg vecuronium) to a myasthenic patient resulted in a
rapid onset of block (lower trace). (From
Baraka A: Onset of neuromuscular block in myasthenic patients. Br J Anaesth 69:227–228,
1992.)
Figure 13-40
Electromyographic response to ulnar nerve stimulation.
Injection of 0.1 mg/kg vecuronium in a normal patient resulted in a slow onset of
nondepolarizing block (upper trace), whereas injecting
one tenth the dose (0.01 mg/kg vecuronium) to a myasthenic patient resulted in a
rapid onset of block (lower trace). (From
Baraka A: Onset of neuromuscular block in myasthenic patients. Br J Anaesth 69:227–228,
1992.)
All myasthenic patients should be closely monitored for neuromuscular weakness postoperatively in the surgical ICU. The differential diagnosis of postoperative weakness in myasthenic patients should include residual effects of neuromuscular blockers or anesthetic drugs, drugs that interfere with neuromuscular transmission (such as aminoglycoside antibiotics, antiarrhythmics, and psychotropics), and myasthenic or cholinergic crisis.
Myotonias are currently differentiated into two different types of disorders. The first type consists of channelopathies ( Fig. 13-36 and Table 13-24 ; also see Table 13-22 ) and includes acquired neuromyotonia, myotonia congenita, paramyotonia, hyperkalemic periodic paralysis, potassium-aggravated myotonia, and hypokalemic periodic paralysis. The latter is a muscle ion channel disorder without myotonia. The second type includes myotonic dystrophy (see the section "Muscular Dystrophies").
Neuromyotonia, also known as Isaacs' syndrome or continuous muscle fiber activity syndrome, is a rare peripheral motor neuron disorder. Like MG and LEMS, neuromyotonia is another example of an acquired (immune mediated) channelopathy. [1077] [1078] [1079] It is believed that autoantibodies target the presynaptic voltage-gated potassium channels (see Fig. 13-36 ), thereby inhibiting action potential repolarization, enhancing transmitter release, and inducing hyperexcitability.[1026] [1077] These antibodies have been detected in neuromyotonia patients.
In contrast to the weaknesses and fatigability seen in MG or LEMS, neuromyotonia is associated with increased activity at the neuromuscular junction that results in severe muscle cramps, stiffness, weakness, and often myokymia (muscular twitching during rest).[1026] Some patients exhibit CNS symptoms such as insomnia, mood changes, and hallucinations.[1077] The association of neuromyotonia with the aforementioned CNS manifestations has been coined "Morvan's syndrome."[1080] A paraneoplastic response often in association with small cell carcinoma or thymoma is seen in about 20% of patients with neuromyotonia.[1081] [1082] Neuromyotonia can coexist with MG.[1018]
Drugs that act by increasing the sodium-pumping action of nerve and muscle tissue, such as phenytoin, are effective in treating neuromyotonia.[1083] Plasmapheresis can provide both clinical and electromyographic improvement.[1084] Immunosuppressive therapy with azathioprine may be helpful in severe cases.
Spinal and epidural anesthesia, as well as succinylcholine and nondepolarizing neuromuscular blockers, are effective in abolishing spontaneous discharge and producing muscle relaxation.[1085] [1086] Peripheral nerve blocks are not effective in abolishing myokymia in all patients, [1086] thus suggesting that in some cases the hyperexcitability originates within the distal nerve trunk.[1079] It should be noted that the abnormal muscle fiber activity can persist during sleep and general anesthesia.
Epidural anesthesia was used successfully for labor and delivery in a patient with neuromyotonia.[1087] The clinical effects of general anesthetics and neuromuscular blockers have not yet been reported in these patients. However, resistance to nondepolarizing neuromuscular blockers is expected in neuromyotonia because of (1) the increased acetylcholine release in these patients[1088] and (2) in vitro evidence of resistance of dTc.[1089] Some patients with acquired neuromyotonia may have autonomic and sensory neuropathies.[1077]
Both dominant (Thomsen) and recessive (Becker) forms of myotonia congenita are caused by mutations in the gene encoding the skeletal muscle voltage-gated chloride channel,[1000] [1001] [1090] with an estimated prevalence of 1 in 23,000 for recessive myotonia and 1 in 50,000 for dominant myotonia.[1013] As discussed before (see the section "The Role of Ion Channels in Neuromuscular Transmission"), chloride channels are responsible for return of the membrane potential to its resting level.[1011] Mutations in this channel decrease Cl- conductance into the cell and thereby lead to hyperexcitability of the muscle membrane and muscle stiffness (see Fig. 13-36 ).[1010] [1011] [1091] Myotonias are characterized by difficulty initiating muscle movement and delayed muscle relaxation after voluntary contraction. It improves with sustained activity (warm-up phenomenon).
Muscle stiffness responds to Na+ channels blockers such as local anesthetics and antiarrhythmic drugs.[1091] Although these drugs do not affect the kinetics of Cl- channels, they decrease cell-membrane excitability.[1092]
Myotonia may be precipitated by cold, shivering, diathermy, succinylcholine, and anticholinesterases (see Table 13-24 ).[945] [1013] [1092] The association between myotonia and malignant hyperthermia is uncertain, probably because of the difficulty in interpretation of the caffeine-halothane contracture test in myotonic patients.[1093] [1094] It is prudent, however, to avoid all anesthetic triggering agents in these patients.
Myotonia developing in response to direct surgical activation of muscle is difficult to prevent and treat.[1095] Unlike local anesthetics and antiarrhythmic drugs, nondepolarizing neuromuscular blockers are not effective in alleviating this myotonic response.[1091] Clinical deterioration may occur in pregnancy and is probably due to the associated hormonal changes. Epidural anesthesia is reported to be safe in these patients. [1096]
Mutations in the skeletal muscle voltage-gated Na+ channel gene produce the clinical phenotypes of hyperkalemic periodic paralysis (HyperPP), paramyotonia congenita, and potassium-aggravated myotonia (see Fig. 13-36 ).[1002] [1012] As discussed before (see the section "The Role of Ion Channels in Neuromuscular Transmission"), voltage-gated sodium channels are responsible for amplification and propagation
|
|
Autosomal Dominant Myotonia Congenita (Thomsen) | Autosomal Recessive Myotonia Congenita (Becker) | Potassium-Aggravated Myotonia | Paramyotonia Congenita | Hyperkalemic Periodic Paralysis (HyperPP) | Hypokalemic Periodic Paralysis (HypoPP) |
|---|---|---|---|---|---|---|
| Age at onset | Infancy-early childhood | Late childhood (variable) | First or second decade | First decade | First decade | Second decade (variable) |
| Initial symptoms | Muscle hypertrophy and generalized myotonia; occasional asymptomatic patients with electrical myotonia | Muscle hypertrophy (legs), generalized myotonia, transient weakness after rest | Myotonia (fluctuans, permanens, painful) affecting facial, eyelid, and paraspinal muscles | Paradoxical eyelid and grip myotonia; focal paralysis common, may overlap with HyperPP | Brief (<1 hr) paralytic attacks; myotonia or paramyotonia (eyelids) between paralytic episodes | Paralytic episodes that last hours-days, tend to remit with age, affect men more than women; no myotonia |
| Provocative stimuli | Myotonia worsened by rest, improves with exercise ("warm-up" phenomenon) | Myotonia worsened by rest or maintenance of same posture ("warm-up") | Potassium, cold, infection, exercise | Exercise and cold | Rest after exercise; cold and potassium trigger both paralysis and myotonia | Rest after exercise, often when waking up in the morning, after carbohydrate-rich and salty meals |
| Myopathy | Weakness may develop in older age; biopsy shows mild abnormalities | Possible muscle atrophy and weakness late in life | Rare myopathy, muscle hypertrophy common | Very rare | Infrequent | Possible progressive myopathy (vacuolar in HypoPP-1, tubular aggregates in HypoPP-2) |
| Therapy | Exercise, antimyotonia therapy (phenytoin, mexiletine) | Exercise, antimyotonia therapy | Acetazolamide, mexiletine, low-potassium diet, flecainide in painful variant | Mild exercise; avoid exposure to cold, mexiletine | Prevention with thiazide diuretics, acetazolamide, sodium restriction, carbohydrate-rich meals; attacks treated with diuretics, calcium gluconate | Prevention with potassium supplements, acetazolamide (worsens HypoPP-2), dichlorophenamide; attacks treated with oral potassium |
| From Kleopa KA, Barchi RL: Genetic disorders of neuromuscular ion channels. Muscle Nerve 26:299–325, 2002. | ||||||
HyperPP is a rare autosomal dominant disorder with a prevalence of 1:100,000.[1012] It is characterized by episodes of muscle weakness associated with hyperkalemia and with signs of myotonia in the interval between attacks.[1010] Respiratory and cardiac muscles are not affected, probably because of the existence of Na+ channels in these muscles different from those expressed in skeletal muscle.[1013] The attacks of paralysis are frequent, brief, and often precipitated by rest after exertion, stress, ingestion of foods with high potassium content such as bananas, or the administration of potassium. A cold environment, emotional stress, and pregnancy provoke or worsen the attacks.[1014] Increases in serum K+ up to 5 to 6 mmol/L may be seen during the attack.[1013] Prophylactic treatment with potassium-wasting diuretics can attenuate the frequency and severity of attacks.
The clinical manifestations of HyperPP, paramyotonia congenita, and potassium-aggravated myotonia are similar, which suggest that the three disorders may be allelic (i.e., a single genetic defect is responsible for coinheritance). [1003] [1004] [1098] [1099] Normokalemic periodic paralysis is a variant of HyperPP and has been reported in only a few families.[1100] [1101] [1102]
Potassium depletion before surgery, maintenance of carbohydrate stores with dextrose-rich, potassium-free intravenous solutions, maintenance of normothermia, and avoidance of acidosis are essential in the anesthetic management of these patients. [1013] [1103] Prewashed packed red blood cells should be used if blood transfusion is required. Careful and frequent monitoring of plasma potassium concentrations and acid-base status is of greatest importance. Succinylcholine should be avoided because it will result in increases in serum potassium concentrations and can cause myotonic symptoms in these patients.[1104] Anticholinesterase drugs should be avoided as well because they may provoke a myotonic reaction.[1105]
An association between malignant hyperthermia and HyperPP in the adult skeletal muscle sodium channel gene has been established.[1106] Patients with HyperPP appear to have a normal response to nondepolarizing neuromuscular blockers.[1103] Hyperkalemia should be considered in the differential diagnosis of postoperative residual weakness. Hyperkalemia should be treated with immediate hyperventilation, calcium chloride, 1.0 to 2.0 mg intravenously, sodium bicarbonate, 1 mEq/kg, and intravenous glucose and insulin (10 U regular insulin in 50 mL 50% glucose or, for children, 0.15 U regular insulin per kilogram in 1.0 mL/kg 50% glucose). Propofol was shown to target and block both normal and mutant voltage-gated sodium channels in a concentration- and voltage-dependent manner.[1107] [1108] Therefore, propofol might be beneficial. Spinal anesthesia is reported to be a safe alternative to general anesthesia in these patients.[1109] Cardiac anesthesia poses particular problems. During recovery, special attention should be directed at maintaining normal body temperature and electrolyte and acid-base status.
Mutations in the skeletal muscle voltage-gated Ca2+ channel gene (DHPR) produce hypokalemic periodic paralysis type 1 (HypoPP-1).[1005] HypoPP-2 is caused by mutations in the gene encoding the voltage-gated Na+ channel of skeletal muscle (see Fig. 13-36 ).[1097] [1098] Both types have the same clinical features.
HypoPP is a rare autosomal dominant disorder with a prevalence of 1:100,000.[1012] It is characterized by episodic weakness associated with hypokalemia during attacks.[1014] The hypokalemia has been attributed to increased activity of the Na+ -K+ pump by insulin, which results in shifting of K+ from the extracellular space into the intracellular compartment.[1111] [1112] As discussed before (see the section "The Role of Ion Channels in Neuromuscular Transmission"), repolarization of the membrane in normal muscle is initiated by the outward K+ current through the potassium ion channel.[1014] The abnormal inward shifting of K+ (into the cell) in HypoPP causes prolonged depolarization leading to inactivation of both the mutant sodium channels in HypoPP-2 and normal sodium channels and thereby results in muscle weakness and paralysis.[1006] [1013] [1113] [1114] Although this description does not include an explicit role for Ca2+ ion channels, the intracellular Ca2+ concentration is essential in regulating insulin secretion,[1115] [1116] and it appears that mutations of the Ca2+ channel in HypoPP-1 may alter this mechanism.[1013] [1112] HypoPP is an example of a loss-of-function mutation of Na+ and Ca2+ ion channels.[1014]
In contrast to HyperPP, myotonia is absent in HypoPP, and ventricular dysrhythmias may occur during hypokalemic attacks.[997] HypoPP attacks are triggered by hypothermia, carbohydrate-rich meals, insulin, and vigorous exercise and can be treated by potassium administration (see Table 13-24 ).[1013] Prophylactic treatment with acetazolamide (a carbonic anhydrase inhibitor) is successful in HypoPP-1 patients, perhaps by producing metabolic acidosis, which decreases the urinary excretion of K+ .[1117] However, acetazolamide should not be used in HypoPP-2 patients because it can induce attacks of weakness and paralysis in this group of patients.[1110]
Preoperative stress should be adequately alleviated by the administration of anxiolytic drugs such as benzodiazepines. Frequent monitoring of plasma potassium concentrations and acid-base status is required.
A normal response to succinylcholine is noted in these patients, [1118] but an association between HypoPP and malignant hyperthermia has been reported.[1119] There are no reports in the literature on the effects of nondepolarizing neuromuscular blockers in these patients, and their use seem to be safe in HypoPP patients.[1118] In a review of 21 anesthetics administered to members of a family with HypoPP, seven patients suffered from mild or severe post-operative paralysis.[1120] Hypokalemia should be considered in the differential diagnosis of postoperative residual weakness.
Spinal anesthesia and epidural anesthesia are safe alternatives to general anesthesia in these patients.[1121] It should be noted, however, that epidural,[1122] axillary, and intercostal nerve blocks[1123] lower serum potassium 0.3 to
|
|
|
|
|
|
|
|
|
|
|
|
|