|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The specific pathways of metabolism (biotransformation) and elimination of neuromuscular blocking drugs are summarized in Table 13-10 . Of the nondepolarizing neuromuscular blockers listed, pancuronium, pipecuronium, vecuronium, atracurium, cisatracurium, mivacurium, and rapacuronium (ORG 9487) are the only ones that are metabolized or degraded. Nearly all nondepolarizing neuromuscular blocker molecules contain ester linkages, acetyl ester groups, and hydroxyl or methoxy groups. These substitutions, especially the quaternary nitrogen
|
|
|
|
Elimination |
|
|
|---|---|---|---|---|---|
| Drug | Duration | Metabolism (%) | Kidney (%) | Liver (%) | Metabolites |
| Succinylcholine | Ultrashort | Butyrylcholinesterase (98%–99%) | <2% | None | Monoester (succinylmonocholine) and choline. The monoester is metabolized much more slowly than succinylcholine is |
| 430A | Ultrashort | Cysteine (fast) and ester hydrolysis (slow) | ? | ? | Inactive cysteine adduction product, chloroformate monoester and alcohol (see Fig. 13-10 ) |
| Mivacurium | Short | Butyrylcholinesterase (95%–99%) | <5% | None | Monoester and quaternary alcohol. The metabolites are inactive. They are most likely not themselves metabolized any further (see Fig. 13-22 ) |
|
|
|
|
(Metabolites eliminated in urine and bile) |
|
|
| Rapacuronium (ORG 9487) | Short | To 3-desacetyl metabolite | 20% | Unknown | The 3-OH derivative, ORG 9488, is two to three times more potent than the parent compound and has a longer half-life |
| Atracurium | Intermediate | Hofmann elimination and nonspecific ester hydrolysis (60%–90%) | 10%–40% | None | Laudanosine, acrylates, alcohols, and acids (see Fig. 13-23 ). Although laudanosine has CNS-stimulating properties, the clinical relevance of this effect is negligible |
|
|
|
|
(Metabolites eliminated in urine and bile) |
|
|
| Cisatracurium | Intermediate | Hofmann elimination (77%?) | Renal clearance is 16% of total |
|
Laudanosine and acrylates. Ester hydrolysis of the quaternary monoacrylate occurs secondarily (see Fig. 13-23 ). Because of the greater potency of cisatracurium, laudanosine quantities produced by Hofmann elimination are 5 to 10 times lower than in the case of atracurium, thus making this a nonissue in practice |
| Vecuronium | Intermediate | Liver (30%–40%) | 40%–50% | 50%–60% | The 3-OH metabolite accumulates, particularly in renal failure. It has about 80% the potency of vecuronium and may be responsible for delayed recovery in ICU patients (see Fig. 13-21 ) |
|
|
|
|
(Metabolites excreted in urine and bile) |
|
|
| Rocuronium | Intermediate | None | <10% | >70% | None |
| Pancuronium | Long | Liver (10%–20%) | 85% | 15% | The 3-OH metabolite may accumulate, particularly in renal failure. It is about two thirds as potent as the parent compound |
| d-Tubocurarine | Long | None | 80% (?) | 20% | None |
| Pipecuronium | Long | Approximately 10% | >90% (?) | <10% | The 3-OH metabolite is produced in small quantities (≅5%) |
| Metocurine | Long | None | >98% | <2% | None |
| Doxacurium | Long | None | >90% (?) | <10% | None |
| Alcuronium | Long | None | 80%–90% (?) | 10%–20% | None |
| Gallamine | Long | None | 100% | 0% | None |
Pancuronium is cleared largely by the kidney.[290] Hepatic uptake of pancuronium is limited.[291] A small amount (15% to 20%) is deacetylated at the 3-position in the liver,[27] [292] [293] but such deacetylation makes a minimal contribution to its total clearance. Deacetylation also occurs at the 17-position, but to such a small extent that it is clinically irrelevant. The metabolites have been individually studied in anesthetized humans. [253] The 3-OH metabolite is the most potent of the three (approximately half the potency of pancuronium) and the only one present in detectable concentrations in plasma. This metabolite
 Figure 13-21
Metabolism of vecuronium as it occurs in the liver.
About 30% to 40% of an injected dose is deacetylated at the 3- and 17-positions.
The major metabolite is 3-OH vecuronium (red arrow).
The metabolites are excreted in urine and bile. The 3-OH metabolite is nearly as
potent as the parent compound and is probably cleared from blood at a rate slightly
slower than that of vecuronium. (Redrawn from Agoston S, Seyr M, Khuenl-Brady
KS, et al: Use of neuromuscular blocking agents in the intensive care unit. Anesthesiol
Clin North Am 11:345, 1993.)
Figure 13-21
Metabolism of vecuronium as it occurs in the liver.
About 30% to 40% of an injected dose is deacetylated at the 3- and 17-positions.
The major metabolite is 3-OH vecuronium (red arrow).
The metabolites are excreted in urine and bile. The 3-OH metabolite is nearly as
potent as the parent compound and is probably cleared from blood at a rate slightly
slower than that of vecuronium. (Redrawn from Agoston S, Seyr M, Khuenl-Brady
KS, et al: Use of neuromuscular blocking agents in the intensive care unit. Anesthesiol
Clin North Am 11:345, 1993.)
Pipecuronium undergoes very little metabolism. Only a very small amount of the drug (5%) may be deacetylated at the 3-position. The major excretory pathway is the kidney, with the liver possibly being a minor secondary pathway. Excretion is delayed, clearance is decreased, and the elimination half-life is lengthened in the presence of major disorders of renal or hepatic function.[293] [298] [299]
Vecuronium, the 2-desmethyl derivative of pancuronium, is more lipid soluble than pancuronium because of absence of the quaternizing methyl group at the 2-position. It undergoes two to three times more metabolism than pancuronium does. Vecuronium is taken up into the liver by a carrier-mediated transport system [291] [300] and is deacetylated at the 3-position by liver microsomes ( Fig. 13-21 ).[301]
The principal metabolite of vecuronium, 3-desacetylvecuronium, is a potent (≅80% of vecuronium) neuromuscular blocking drug in its own right. The metabolite, though, has lower plasma clearance and a longer duration of action than vecuronium does.[302] 3-Desacetylvecuronium has a clearance of 3.5 mL/kg/min, and renal clearance accounts for approximately a sixth of its elimination.[302] In patients in the ICU who have renal failure, 3-desacetylvecuronium can accumulate and produce prolonged neuromuscular blockade.[305] Other putative metabolites are 17-desacetylvecuronium and 3,17-bisdesacetylvecuronium, neither of which occurs in clinically significant amounts.[27]
Rocuronium is eliminated primarily by the liver,[306] [307] with a small fraction (≅10%) eliminated in urine.[308] It is taken up into the liver by a carrier-mediated active transport system.[309] [310] The putative metabolite
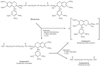 Figure 13-22
Metabolism of mivacurium by butyrylcholinesterase. The
reaction occurs at about 70% to 88% of the rate of succinylcholine in vitro. The
metabolites are inactive and carry positive charges, thus suggesting minimal central
nervous system entry. (Redrawn from Savarese JJ, Ali HH, Basta SJ, et al:
The clinical neuromuscular pharmacology of mivacurium chloride [BW B1090U]. A short-acting
nondepolarizing ester neuromuscular blocking drug. Anesthesiology 68:723–732,
1988.)
Figure 13-22
Metabolism of mivacurium by butyrylcholinesterase. The
reaction occurs at about 70% to 88% of the rate of succinylcholine in vitro. The
metabolites are inactive and carry positive charges, thus suggesting minimal central
nervous system entry. (Redrawn from Savarese JJ, Ali HH, Basta SJ, et al:
The clinical neuromuscular pharmacology of mivacurium chloride [BW B1090U]. A short-acting
nondepolarizing ester neuromuscular blocking drug. Anesthesiology 68:723–732,
1988.)
Rapacuronium has a clearance of between 8 and 11 mL/kg/min.[219] [311] It probably undergoes metabolism to its 3-desacetyl derivative (ORG 9488), which contributes significantly to its neuromuscular blocking effect.[311] The metabolite (ORG 9488) is more potent and shows considerably slower elimination than does the parent compound. Accumulation of the metabolite may be the reason for slower recovery after successive doses of rapacuronium.[311]
Mivacurium is hydrolyzed in plasma by butyryl-cholinesterase to monoester and the amino alcohol ( Fig. 13-22 ).[10] [312] These compounds are excreted in urine and bile.[313] The metabolites are positively charged, thus making central nervous system (CNS) entry unlikely. They show less than 1/100 the neuromuscular blocking activity of the parent compound. They do not affect the autonomic nervous system.[313] Less than 5% is excreted as the parent compound in urine.
Mivacurium consists of three stereoisomers, and clearance of the two most pharmacologically active isomers, the cis-trans and trans-trans isomers, is approximately 100 and 50 to 70 mL/kg/min, respectively.[171] [172] [314] These two isomers have elimination half-lives of 2 to 3 minutes.[171] The third stereoisomer, the cis-cis isomer, is present as only 4% to 8% of the mivacurium mixture and has less than 10% of the neuromuscular blocking potency of the other two isomers.[171] Consequently, even though it has a much longer elimination half-life (55 minutes) and lower clearance (≅4 mL/kg/min) than the two other isomers, it does not contribute significantly to the duration of action of mivacurium.[171] This rapid enzymatic clearance of mivacurium accounts for its short duration.[10] [171] Mivacurium has a duration of action much shorter than that of vecuronium and atracurium but about twice that of succinylcholine.[315]
When butyrylcholinesterase activity is severely deficient, such as in rare patients (1/3000) who are homozygotes with genetically atypical enzyme, the duration of action of mivacurium is prolonged for up to several hours.[316] [317] [318] [319] [320]
Theoretically, atracurium is metabolized through two pathways (see Fig 13-23 ).[321] The drug undergoes Hofmann elimination and ester hydrolysis by nonspecific esterases. Hofmann elimination is a purely chemical process that results in loss of the positive charges by molecular fragmentation to laudanosine (a tertiary amine) and a mono-quaternary acrylate.[322] [323] They were thought to have no neuromuscular and little or no cardiovascular activity of clinical relevance.[322] [323] Under the proper chemical conditions, these breakdown products may actually be used to synthesize the parent compound.
Because it undergoes Hofmann elimination, atracurium is relatively stable at pH 3.0 and 4°C and becomes unstable when injected into the bloodstream. Early observations of breakdown of the drug in buffer and plasma showed faster degradation in plasma, thus suggesting possible enzymatic hydrolysis of the ester groups.[324] Further evidence suggests that this second pathway, ester hydrolysis, may be of more importance than was originally realized in the breakdown of atracurium.[325] Through the use of pharmacokinetic analysis, Fisher and associates[326] concluded that a significant amount of clearance of atracurium may be accomplished by routes other than ester hydrolysis and Hofmann elimination. Atracurium's metabolism is complicated and may not be completely resolved.[326] [327]
Laudanosine, a metabolite of atracurium, has CNS-stimulating properties. Unlike atracurium, laudanosine is dependent on the liver and kidney for elimination and has a long elimination half-life.[328] [329] Laudanosine concentrations are elevated in patients with liver disease[330] and those who have received atracurium for many hours in an ICU.[331] Laudanosine freely crosses the blood-brain barrier.[328] Beemer and coworkers[332] found that patients awakened at a 20% higher arterial concentration of thiopental when atracurium had been given; such awakening was attributed to the CNS stimulatory effect of laudanosine. These relatively low concentrations of laudanosine, however, did not influence animal models of epilepsy[333] or lidocaine-induced seizures. [334] In the ICU, blood levels of laudanosine can be as high as 5.0 to 6.0 µg/mL.[331] Though not known in humans, the seizure threshold in animals ranges from 5.0 µg/mL in rabbits[335] to 17 µg/mL in dogs.[336] Thus, adverse effects are unlikely to occur with atracurium use in the operating room or the ICU. Laudanosine also has cardiovascular effects. In dogs, hypotension occurs at a blood concentration of about 6 µg/mL,[328] [336] a level higher than usually found in patients in the ICU. However, there is one case report of a patient who had severe hypotension and bradycardia while receiving atracurium, which resolved only when vecuronium was substituted.[337] Laudanosine enhances stimulation-evoked release of norepinephrine,[338] [339] a characteristic that may also partly account for its CNS-stimulating effect.
Atracurium is a mixture of 10 optical isomers. Cisatracurium is the 1R cis-1'R cis isomer of atracurium.[167] Like atracurium, it is metabolized by Hofmann elimination to laudanosine and a monoquaternary alcohol metabolite.[340] [341] [342] There is no ester hydrolysis of the parent molecule.[340] Hofmann elimination accounts for 77% of the total clearance of 5 to 6 mL/kg/min.[343] Twenty-three percent of the drug is cleared through organ-dependent means, with renal elimination accounting for 16% of this total.[342] Because cisatracurium is about four to five times as potent as atracurium, about five times less laudanosine is produced, and accumulation of this metabolite is not thought to be of any consequence in clinical practice.
dTc does not undergo active metabolism. The kidney is the major pathway of elimination, with approximately 50% of a dose being eliminated through renal pathways. The liver is probably a secondary route of elimination. The drug is not indicated for use in patients with either renal[234] or hepatic[344] failure because more suitable agents are available.
Metocurine is excreted only by the kidney. It has no alternative biliary pathway, and no metabolism occurs in the liver.[345]
Doxacurium is not metabolized in humans. The drug is excreted in urine and bile as the unchanged parent molecule. Urine is the major route of elimination, with bile being a minor secondary pathway.[346]
430A appears to be degraded by two chemical mechanisms, neither of which is enzymatic: (1) rapid formation of an apparently inactive cysteine adduction product, with cysteine replacing chlorine, and (2) slower hydrolysis of the ester bond adjacent to the chlorine substitution to presumably inactive hydrolysis products (see Fig. 13-10 ).[187]
Gallamine is not metabolized and is excreted unchanged in the urine only. It does not have any alternative biliary excretory pathway.[347]
Alcuronium undergoes little or no metabolism. Urine is the major excretory pathway, with a small amount of biliary clearance of unchanged drug.[190]
In summary, the long-acting neuromuscular blockers undergo minimal or no metabolism, and they are
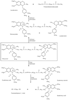 Figure 13-23
Degradation pathways of atracurium. The major metabolite,
laudanosine, is excreted in urine and bile. Laudanosine is a tertiary amine that
may enter the central nervous system. Less than 10% of atracurium is excreted as
the parent compound. (Redrawn from Basta SJ, Ali HH, Savarese JJ, et al:
Clinical pharmacology of atracurium besylate [BW 33A]: A new non-depolarizing muscle
relaxant. Anesth Analg 61:723–729, 1982.)
Figure 13-23
Degradation pathways of atracurium. The major metabolite,
laudanosine, is excreted in urine and bile. Laudanosine is a tertiary amine that
may enter the central nervous system. Less than 10% of atracurium is excreted as
the parent compound. (Redrawn from Basta SJ, Ali HH, Savarese JJ, et al:
Clinical pharmacology of atracurium besylate [BW 33A]: A new non-depolarizing muscle
relaxant. Anesth Analg 61:723–729, 1982.)
Neuromuscular blockers of intermediate duration such as vecuronium, rocuronium, atracurium, and cisatracurium have clearances in the range 3 to 6 mL/kg/min because of multiple pathways of degradation, metabolism, and elimination. Atracurium is cleared two to three times more rapidly than the long-acting drugs are.[227] [330] [348] [349] [350] [351] Similar clearance values have been obtained for rocuronium[352] [353] [354] [355] [356] [357] and cisatracurium. [341] [342] [343] [358] [359]
The only short-acting neuromuscular blocker currently available for clinical use, mivacurium, is cleared rapidly and almost exclusively through metabolism by butyrylcholinesterase, which results in plasma clearance much greater than that of any other nondepolarizing neuromuscular blocker.[10]
|
|
|
|
|
|
|
|
|
|
|
|
|