|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The use of neuromuscular blocking drugs in anesthesia has its origin in the South American Indians' arrow poisons or curares. Several nondepolarizing neuromuscular blockers are still purified from naturally occurring sources. For example, although dTc can be synthesized, it is still less expensive to isolate it from the Amazonian vine Chondodendron tomentosum. Similarly, the intermediates for the production of metocurine and alcuronium, which are semisynthetic, are obtained from Chondodendron and Strychnos toxifera. Malouetine, the first steroidal neuromuscular blocking drug, was originally isolated from Malouetia bequaertiana, which grows in the jungles of Zaire in central Africa. Pancuronium, vecuronium, pipecuronium, rocuronium, rapacuronium, atracurium, doxacurium, mivacurium, cisatracurium, and gallamine are entirely synthetic.
The available nondepolarizing neuromuscular blockers can be classified according to chemical class (steroidal, benzylisoquinolinium, or other compounds) or according to onset or duration of action (long-, intermediate-, and short-acting drugs) of equipotent doses ( Table 13-4 ).
Nondepolarizing neuromuscular blocking drugs were originally classified by Bovet[61] as pachycurares, or bulky molecules having the amine functions incorporated into rigid ring structures. Two extensively studied chemical series of synthetic nondepolarizing neuromuscular blockers are (1) the aminosteroids (steroidal), in which the distance is maintained by an androstane skeleton, and (2) the benzylisoquinolinium series, in which the distance is maintained by linear diester-containing chains or, in the case of curare, by benzyl ethers. For a detailed account of structure-activity relationships, see Lee.[159]
dTc is a neuromuscular blocker in which the amines are present in the form of two benzyl-substituted tetrahydroisoquinoline structures ( Fig. 13-7 ). The quaternary/tertiary nature of the two amines was initially questioned; however, in nuclear magnetic resonance spectroscopy and methylation/demethylation studies, Everett and colleagues[160] demonstrated that dTc contains only three N-methyl groups. One amine is quaternary (permanently charged with four nitrogen substituents) and the other tertiary (pH-dependent charge with three nitrogen substituents). At physiologic pH, the tertiary
|
|
Clinical Duration | |||
|---|---|---|---|---|
|
|
Long Acting (>50 min) | Intermediate Acting (20–50 min) | Short Acting (10–20 min) | Ultrashort Acting (<10 min) |
| Steroidal compounds | Pancuronium | Vecuronium | Rapacuronium |
|
|
|
Pipecuronium | Rocuronium |
|
|
| Benzylisoquinolinium compounds | d-Tubocurarine | Atracurium | Mivacurium |
|
|
|
Metocurine | Cisatracurium |
|
|
|
|
Doxacurium |
|
|
|
| Others |
|
|
|
|
| Asymmetric mixed-onium chlorofumarates |
|
|
|
430A |
| Bisquaternary tropinyl diester |
|
|
|
TAAC3 |
| Phenolic ether | Gallamine |
|
|
|
| Diallyl derivative of toxiferine | Alcuronium |
|
|
|
| Most nondepolarizing neuromuscular blockers are bisquaternary ammonium compounds. d-Tubocurarine, vecuronium, rocuronium, and rapacuronium are monoquaternary compounds, and gallamine is a trisquaternary ammonium compound. | ||||
 Figure 13-7
Chemical structure of d-tubocurarine,
metocurine, and chondocurine.
Figure 13-7
Chemical structure of d-tubocurarine,
metocurine, and chondocurine.
Atracurium is a bis-benzyltetrahydroisoquinolinium with isoquinolinium nitrogens connected by a diester-containing hydrocarbon chain ( Fig. 13-8 ). The presence (in duplicate) of two-carbon separations between quaternary nitrogen and ester carbonyl provides the substrate for a Hofmann degradation reaction. [9] [164] In a Hofmann elimination reaction, a quaternary ammonium group is converted into a tertiary amine by cleavage of a carbon-nitrogen bond. This reaction is pH and temperature dependent, with higher pH and temperature favoring elimination. The actual structure of the quaternary centers is the laudanosinium moiety as in metocurine. Atracurium has four chiral centers at each of the adjacent chiral carbons of the two amines. The marketed product has 10 isomers.[164] [165] These isomers have been separated into three geometric isomer groups that are designated cis-cis, cis-trans, and trans-trans based on their configuration about the tetrahydroisoquinoline ring system.[164] [165] The ratio of the cis-cis, cis-trans, and trans-trans isomers is
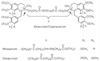 Figure 13-8
Chemical structure of atracurium, cisatracurium, mivacurium,
and doxacurium. *Chiral centers arrows showing
the cleavage sites for Hofmann elimination.
Figure 13-8
Chemical structure of atracurium, cisatracurium, mivacurium,
and doxacurium. *Chiral centers arrows showing
the cleavage sites for Hofmann elimination.
Cisatracurium is the 1R cis-1'R cis isomer of atracurium and represents about 15% of the marketed atracurium mixture by weight, but more than 50% in terms of potency or neuromuscular blocking activity (see Fig. 13-8 ). R designates the absolute stereochemistry of benzyltetrahydroisoquinoline rings, and cis represents the relative geometry of the bulky dimethoxy and 2-alkyester groups at C(1) and N(1), respectively.[167] [168] Cisatracurium is metabolized by Hofmann elimination. It is approximately four times as potent as atracurium, and unlike atracurium, it does not cause histamine release in the clinical dose range.[167] [169] This observation indicates that the phenomenon of histamine release may be stereospecific.[167] [170] Cisatracurium is the second benzylisoquinolinium (after doxacurium) to be largely free of this side effect.
Mivacurium differs from atracurium by the presence of an additional methylated phenolic group (see Fig. 13-8 ). When compared with other isoquinolinium neuromuscular blockers, the interonium chain of mivacurium is longer (16 atoms).[162] Mivacurium consists of a mixture of three stereoisomers.[171] The two most active are the trans-trans and cis-trans isomers (57% and 37% weight per weight [w/w], respectively), which are equipotent; the cis-cis isomer (6% w/w) has only a tenth the activity of the others in cats and monkeys.[171] Mivacurium is metabolized by butyrylcholinesterase at about 70% to 88% the rate of succinylcholine to a monoester, a dicarboxylic acid.[10] [172]
Doxacurium is a bisquaternary benzylisoquinolinium diester of succinic acid (see Fig. 13-8 ). The interonium chain is shorter than that in either atracurium or mivacurium. Lee [159] pointed out that the number of methoxy groups on benzylisoquinolinium heads is increased from four (atracurium) and five (mivacurium) to six (doxacurium).[162] This increase was associated with both an increase in potency and a reduction in the propensity to release histamine. [159] [162]
In the steroidal compounds, it is probably essential that one of two nitrogen atoms in the molecule be quaternized.[173] The presence of acetyl ester (acetylcholine-like moiety) is thought to facilitate its interaction with nAChRs at the postsynaptic muscle membrane.[27] [174]
Pancuronium is characterized by the presence of two acetyl ester groups on the A and D rings of the steroidal molecule. Pancuronium is a potent neuromuscular blocking drug with both vagolytic and butyrylcholinesterase-inhibiting properties ( Fig. 13-9 ).[175] Deacetylation of the 3-OH or 17-OH groups decreases its potency.[176]
Vecuronium is an N-demethylated derivative of pancuronium in which the 2-piperidine substituent is not methylated (vecuronium lacks the N-methyl group at position 2) (see Fig. 13-9 ).[7] [27] At physiologic pH, the tertiary amine is largely protonated similar to dTc. The minor molecular modification in comparison to pancuronium resulted in (1) a slight change in the potency; (2) a marked reduction in vagolytic properties; (3) molecular instability in solution, which explains in part the shorter duration of action of vecuronium than pancuronium; and (4) increased lipid solubility, which results in greater biliary elimination of vecuronium than pancuronium.[27] [162]
Pancuronium and vecuronium are very similar in structure, yet vecuronium is prepared as a lyophilized powder. Vecuronium is degraded by the hydrolysis of either (or both) acetyl esters at the C3- and C17-positions. Hydrolysis at the C3-position is the primary degradation product. The acetate at the 3-position is more susceptible to hydrolysis in aqueous solutions. Vecuronium is less stable in solution because the group effect of the adjacent basic piperidine at the 2-position facilitates hydrolysis of the 3-acetate. Therefore, vecuronium cannot be prepared as a ready-to-use solution with a sufficient shelf life, even as a buffered solution. [177] In pancuronium, the 2-piperidine is quaternized and no longer basic. Thus, it does not participate in catalysis of the 3-acetate hydrolysis.[177]
Pipecuronium, like pancuronium, is a bisquaternary compound. Pipecuronium has piperazine rings attached to the A and D rings of the steroid nucleus, whereas pancuronium has piperidine rings (see Fig. 13-9 ).[162] Pipecuronium is a nonvagolytic substitute for pancuronium. Changes in the quaternary groups, in which the quaternary nitrogen atoms were placed at the distal (4-position) aspect of the 2,16-β substitutions, lessen the vagolytic effects.[162] As a result, pipecuronium is about 10 times less vagolytic than pancuronium.
Rocuronium lacks the acetyl ester that is found in the steroid nucleus of pancuronium and vecuronium in the A ring (see Fig. 13-9 ). The introduction of cyclic substituents
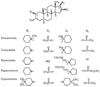 Figure 13-9
Chemical structure of different steroidal neuromuscular
blockers.
Figure 13-9
Chemical structure of different steroidal neuromuscular
blockers.
Rapacuronium became available in the United States in 1999 but was withdrawn from the market by the manufacturer in spring 2001 because of a high incidence of respiratory complications.[181] [182] [183] [184] Rapacuronium is a monoquaternary compound that has the same basic steroid backbone as the rest of the steroidal neuromuscular blockers (see Fig. 13-9 ).
The agent 430A ( Fig. 13-10 ) represents a new class of non-depolarizing neuromuscular blockers called asymmetric mixed-onium chlorofumarates. The presence of three methyl groups between the quaternary nitrogen and oxygen atom at each end of the carbon chain suggests that similar to mivacurium, this compound will not undergo Hofmann degradation.[13] [185] [186] [187] The compound has an ultrashort duration of action in human volunteers and different animal species. A study in anesthetized human volunteers evaluated the onset and recovery profiles of 430A in the thumb and larynx. The pattern of blockade resembles that of succinylcholine, with fully paralyzing doses (2 to 3 × ED95 or 0.38 to 0.54 mg/kg) producing 100% block of TOF stimulation within 50 to 60 seconds in the larynx. Spontaneous recovery to a TOF of 0.9 develops in the thumb within about 12 to 15 minutes after the administration of doses as large as 0.54 mg/kg (or 3 × ED95 ). Recovery is accelerated by edrophonium. The cardiovascular changes after rapid (5 second) bolus doses in these volunteers have averaged less than 10% from baseline.[188]
TAAC3 is a bis[N-(3,4-diacetoxybenzyl)tropanium-3α-yl] glutarate dibromide ( Fig. 13-11 ). [12] No data on this compound in humans have been published. In animals, TAAC3 has a slower onset and shorter duration than succinylcholine has.[189] The short duration of action of TAAC3 is attributed to enzymatic hydrolysis of the acetoxy groups on the quaternary benzyl groups by nonspecific carboxyesterases.[189] At ED90 doses, TAAC3 exhibits a moderate degree of cardiac vagal block, and in large doses of 5 to 10 × ED90 , it produces significant hypotension in the dog that is not related to histamine release.[189] The cardiac vagal blocking effect of TAAC3 in equipotent doses is similar to that of rocuronium.[189]
Gallamine is a trisquaternary substance ( Fig. 13-12 ). Its potent vagolytic activity is due to the presence of three positively charged nitrogen atoms. Gallamine was synthesized originally by Bovet [61] as part of an extensive structure-activity study that helped evolve the concepts of "pachycurares" and "leptocurares." Succinylcholine also evolved from this work, for which Bovet received the Nobel Prize.
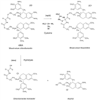 Figure 13-10
Chemical structure of 430A (a mixed-onium chlorofumarate).
In whole human blood, two pathways of deactivation occur, neither of which is enzymatic:
(1) rapid formation of an apparently inactive cysteine adduction product with cysteine
replacing chlorine and (2) slower hydrolysis of the ester bond adjacent to the chlorine
substitution to chlorofumarate monoester and alcohol.[187]
Figure 13-10
Chemical structure of 430A (a mixed-onium chlorofumarate).
In whole human blood, two pathways of deactivation occur, neither of which is enzymatic:
(1) rapid formation of an apparently inactive cysteine adduction product with cysteine
replacing chlorine and (2) slower hydrolysis of the ester bond adjacent to the chlorine
substitution to chlorofumarate monoester and alcohol.[187]
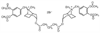 Figure 13-11
Chemical structure of TAAC3.
Figure 13-11
Chemical structure of TAAC3.
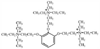 Figure 13-12
Chemical structure of gallamine, a trisquaternary ether
of gallic acid. Gallamine is the only trisquaternary compound available. Its strong
vagolytic property is probably due to the trisquaternary structure.
Figure 13-12
Chemical structure of gallamine, a trisquaternary ether
of gallic acid. Gallamine is the only trisquaternary compound available. Its strong
vagolytic property is probably due to the trisquaternary structure.
Introduced in 1964, alcuronium is a long-acting drug that is the semisynthetic diallyl derivative of toxiferine ( Fig. 13-13 ). The latter is purified from Strychnos toxifera. Its advantage at the time of its introduction was a relative lack of side effects. It is midly vagolytic and is excreted unchanged by the kidney with a minor secondary biliary pathway. Alcuronium is moderately popular in Europe, the Far East, and Australia, but it is not available in the United States.
|
|
|
|
|
|
|
|
|
|
|
|
|