|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Morphine pharmacokinetics are notably different from those of
the fentanyl congeners. This difference is in
|
|
Morphine | Meperidine | Fentanyl | Sufentanil | Alfentanil | Remifentanil |
|---|---|---|---|---|---|---|
| pKa | 8.0 | 8.5 | 8.4 | 8.0 | 6.5 | 7.1 |
| % Un-ionized at pH 7.4 | 23 | <10 | <10 | 20 | 90 | 67? |
| Octanol/H2 O partition coefficient | 1.4 | 39 | 813 | 1,778 | 145 | 17.9 |
| % Bound to plasma protein | 20–40 | 39 | 84 | 93 | 92 | 80? |
| Diffusible fraction (%) | 16.8 | 2.2 | 1.5 | 1.6 | 8.0 | 13.3? |
| t½α (min) | 1–2.5 | — | 1–2 | 1–2 | 1–3 | 0.5–1.5 |
| t½β (min) | 10–20 | 5–15 | 10–30 | 15–20 | 4–17 | 5–8 |
| t½γ (hr) | 2–4 | 3–5 | 2–4 | 2–3 | 1–2 | 0.7–1.2 |
| Vdc (L/kg) | 0.1–0.4 | 1–2 | 0.4–1.0 | 0.2 | 0.1–0.3 | 0.06–0.08 |
| Vdss (L/kg) | 3–5 | 3–5 | 3–5 | 2.5–3.0 | 0.4–1.0 | 0.2–0.3 |
| Clearance (mL/min/kg) | 15–30 | 8.18 | 10–20 | 10–15 | 4–9 | 30–40 |
| Hepatic extraction ratio | 0.6–0.8 | 0.5–0.7 | 0.8–1.0 | 0.7–0.9 | 0.3–0.5 | NA |
| t½ α, β, γ are the half-lives of a three-compartment model; Vdc , volume of distribution of the central compartment; Vdss , volume of distribution at steady state. | ||||||
| From Bailey PL, Egan TD, Stanley TH: Intravenous opioid anesthetics. In Miller RD (ed): Anesthesia, 5th ed. New York, Churchill Livingstone, 2000, p 312. | ||||||
The pKa of morphine (8.0) is greater than physiologic pH, and thus, after intravenous injection only a small fraction (10% to 20%) of morphine is un-ionized. Penetration of morphine into and out of the brain is presumably slower compared with that of other opioids. Approximately 20% to 40% of morphine is bound to plasma proteins, mostly albumin.
Morphine is principally metabolized by conjugation in the liver, but the kidney plays a key role in the extrahepatic metabolism of morphine.[298] [299] Morphine 3-glucuronide (M3G) is the major metabolite of morphine, but it does not bind to opioid receptors and possesses little or no
Unlike morphine, after intravenous injection first-pass uptake of meperidine by the lungs is approximately 65%.[296] Meperidine is more highly bound to plasma proteins than morphine is, principally (70%) to α1 -acid glycoprotein. As with morphine, a relatively high hepatic extraction ratio results in biotransformation that is dependent on hepatic blood flow. The major metabolite normeperidine has analgesic activity and is roughly twice as potent as meperidine in producing seizures in animals. The elimination half-life of normeperidine is considerably longer than that of meperidine, and thus repeated doses can easily produce accumulation of this toxic metabolite in patients with renal disease, potentially producing seizures.
A three-compartment model is typically used to describe plasma fentanyl concentration decay. The lungs exert a significant first-pass effect and transiently take up approximately 75% of an injected dose of fentanyl.[296] [305] Approximately 80% of fentanyl is bound to plasma proteins, and significant amounts (40%) are taken up by red blood cells.[306] Fentanyl has a relatively long half-life, in large part because of this widespread distribution (i.e., large volume of distribution) in body tissues.
Fentanyl is primarily metabolized in the liver by N-dealkylation and hydroxylation. Metabolites begin to appear in the plasma as early as 1.5 minutes after injection. In humans, norfentanyl, the primary metabolite, is detectable in the urine for up to 48 hours after intravenous administration of fentanyl.
Following intravenous injection, alfentanil plasma concentrations are described by either two-compartment[307] or three-compartment models.[308] [309] Alfentanil is bound to plasma proteins (mostly glycoproteins) in higher
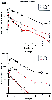 Figure 11-13
Mean plasma concentrations (± SEM) of morphine,
morphine-6-glucuronide (M-6-G), and morphine-3-glucuronide (M-3-G) after intravenous
and oral administration of morphine. (From Osborne R, Joel S, Trew D, Slevin
M: Morphine and metabolite behavior after different routes of morphine administration:
demonstration of the importance of the active metabolite morphine-6-glucuronide.
Clin Pharmacol Ther 47:12–9, 1990.)
Figure 11-13
Mean plasma concentrations (± SEM) of morphine,
morphine-6-glucuronide (M-6-G), and morphine-3-glucuronide (M-3-G) after intravenous
and oral administration of morphine. (From Osborne R, Joel S, Trew D, Slevin
M: Morphine and metabolite behavior after different routes of morphine administration:
demonstration of the importance of the active metabolite morphine-6-glucuronide.
Clin Pharmacol Ther 47:12–9, 1990.)
The main metabolic pathways of alfentanil are similar to those of sufentanil and include oxidative N-dealkylation and O-demethylation, aromatic hydroxylation, and ether
Until recently, the pharmacokinetics of sufentanil had been inadequately characterized because of poor assay sensitivity.[312] Sufentanil's potency is so great that it continues to exert its effects when the concentrations in the plasma are at very low levels. Therefore, it was necessary to develop an assay that could measure sufentanil at low levels in plasma.
The pharmacokinetic properties of sufentanil are adequately described by a three-compartment model.[313] After intravenous injection, first-pass pulmonary extraction, retention, and release are similar to those of fentanyl.[297] The pKa of sufentanil at physiologic pH is the same as that of morphine (8.0) and, therefore, only a small amount (20%) exists in the un-ionized form. Sufentanil is twice as lipid-soluble as fentanyl and is highly bound (93%) to plasma proteins including α1 -acid glycoprotein. The major metabolic pathways of sufentanil include N-dealkylation, oxidative O-demethylation, and aromatic hydroxylation. [314] Major metabolites include N-phenylpropanamide.
Although chemically related to the fentanyl congeners, remifentanil is structurally unique because of its ester linkages. Remifentanil's ester structure renders it susceptible to hydrolysis by blood- and tissue-nonspecific esterases, resulting in rapid metabolism. Remifentanil thus constitutes the first "ultrashort"-acting opioid for use as a supplement to general anesthesia.
The pharmacokinetic properties of remifentanil are best described by a three-compartment model.[76] [315] [316] Its clearance is several times greater than that of normal hepatic blood flow, consistent with widespread extrahepatic metabolism. However, remifentanil is not significantly metabolized or sequestered in the lungs. [317] It is a weak base with a pKa of 7.07. It is highly lipid-soluble with an octanol/water partition coefficient of 19.9 at pH 7.4. Remifentanil is highly bound (70%) to plasma proteins (mostly α1 -acid glycoprotein). The remifentanil free base is formulated as a solution with glycine. Because glycine has been shown to act as an inhibitory neurotransmitter that causes a reversible motor weakness when injected intrathecally in rodents, remifentanil is not approved for spinal or epidural use.[318]
The primary metabolic pathway of remifentanil is de-esterification to form a carboxylic acid metabolite, GI90291 ( Fig. 11-14 ),[319] which is 0.001 to 0.003 times as potent as remifentanil. The low in vivo potency of GI90291 can be explained by a low affinity to the μ-receptor in combination with poor brain penetration.[320] Excretion of GI90291 is dependent on renal clearance mechanisms. Evidence from studies in dogs suggests that the remifentanil metabolites are, for practical purposes, completely inactive, even in the face of renal failure. Its pharmacokinetics are not appreciably influenced by renal or hepatic failure.[321] [322] In blood, remifentanil is
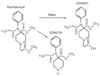 Figure 11-14
Metabolic pathway of remifentanil. De-esterification
by nonspecific plasma and tissue esterases to form a carboxylic acid metabolite (GI90291)
that has only 1/300 to 1/1,000 the potency of the parent compound is the primary
metabolic pathway. N-dealkylation of remifentanil
to GI94219 is a minor metabolic pathway. (From Egan TD, Lemmens HJ, Fiset
P, et al: The pharmacokinetics of the new short-acting opioid remifentanil [GI87084B]
in healthy adult male volunteers. Anesthesiology 79:881–892, 1993.)
Figure 11-14
Metabolic pathway of remifentanil. De-esterification
by nonspecific plasma and tissue esterases to form a carboxylic acid metabolite (GI90291)
that has only 1/300 to 1/1,000 the potency of the parent compound is the primary
metabolic pathway. N-dealkylation of remifentanil
to GI94219 is a minor metabolic pathway. (From Egan TD, Lemmens HJ, Fiset
P, et al: The pharmacokinetics of the new short-acting opioid remifentanil [GI87084B]
in healthy adult male volunteers. Anesthesiology 79:881–892, 1993.)
|
|
|
|
|
|
|
|
|
|
|
|
|