Common Pharmacokinetic Features of Opioids
Representative pharmacokinetic parameters for the opioids commonly
used in anesthesia are displayed in Table
11-7
.
After intravenous injection, arterial plasma concentrations of
opioids rise to a peak within one circulation time. Thereafter, they exhibit a rapid
redistribution phase and a slower elimination phase typical of drugs whose pharmacokinetics
are described by compartmental models.[295]
 Figure 11-11
Pharmacokinetic and pharmacodynamic variability of the
opioid. The upper panel demonstrates alfentanil
plasma concentration in 45 patients administered with 50 µg/kg alfentanil.
The lower panel shows dose-response relationships
of alfentanil from 34 patients receiving alfentanil and 66% nitrous oxide during
intraabdominal surgery. The black circles represent
the Cp50
from each patient. Cp50
, the concentration needed
to obtund responses to stimuli adequately in 50% of patients. (From Maitre
PO, Vozeh S, Heykants J, et al: Population pharmacokinetics of alfentanil: The
average dose-plasma concentration relationship and interindividual variability in
patients. Anesthesiology 66:3–12, 1987; and Ausems ME, Hug CC Jr, Stanski
DR, Burm AG: Plasma concentrations of alfentanil required to supplement nitrous
oxide anesthesia for general surgery. Anesthesiology 65:362–367, 1986.)
Figure 11-11
Pharmacokinetic and pharmacodynamic variability of the
opioid. The upper panel demonstrates alfentanil
plasma concentration in 45 patients administered with 50 µg/kg alfentanil.
The lower panel shows dose-response relationships
of alfentanil from 34 patients receiving alfentanil and 66% nitrous oxide during
intraabdominal surgery. The black circles represent
the Cp50
from each patient. Cp50
, the concentration needed
to obtund responses to stimuli adequately in 50% of patients. (From Maitre
PO, Vozeh S, Heykants J, et al: Population pharmacokinetics of alfentanil: The
average dose-plasma concentration relationship and interindividual variability in
patients. Anesthesiology 66:3–12, 1987; and Ausems ME, Hug CC Jr, Stanski
DR, Burm AG: Plasma concentrations of alfentanil required to supplement nitrous
oxide anesthesia for general surgery. Anesthesiology 65:362–367, 1986.)
After administration into a central compartment, opioids are either
eliminated from the central compartment (by excretion or biotransformation) or distributed
to peripheral compartments. In general, opioids are cleared from the plasma by biotransformation
in the liver. However, extrahepatic metabolism is important for some opioids.
As a rule, because of their high lipid solubility, opioids are
widely distributed in body tissues. In pharmacokinetic terms, this means that opioids
typically have a high apparent volume of distribution at steady state. Because opioids
are distributed so widely and so rapidly to various body tissues, redistribution
has a prominent impact on
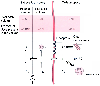 Figure 11-12
Distribution of fentanyl within the body compartments
at the extracellular pH of 7.4. The accumulation of fentanyl by the tissue is assumed
to be 13-fold. In the extracellular space, an equilibrium exists between the ionized
fentanyl (F+
), the free base (F), and the fentanyl molecules bound to
macromolecules (Fb
). The concentration of ionized fentanyl within the
interstitial fluid (encircled F+
) determines the pharmacologic effect,
because the opioid receptors are located at the cell surface. Fentanyl as free base
readily penetrates into the cells and becomes bound to cytomembranes, lysosomes,
and other structures (thick arrows) and thus accumulates
in the cell. A small decrease of the extracellular pH will shift the equilibrium
between F and F+
toward higher F+
concentrations and will induce
a marked release of fentanyl from the cellular compartment as a result of the decrease
of the concentration of F in the interstitium. An increase of the extracellular
H+
concentration by pH 0.2 might roughly double the concentration of ionized
fentanyl within the interstitial fluid and accordingly enhance its pharmacologic
effect. (From Lullmann H, Martins BS, Peters T: pH-dependent accumulation
of fentanyl, lofentanil and alfentanil by beating guinea pig atria. Br J Anaesth
57:1012–1017, 1985.)
Figure 11-12
Distribution of fentanyl within the body compartments
at the extracellular pH of 7.4. The accumulation of fentanyl by the tissue is assumed
to be 13-fold. In the extracellular space, an equilibrium exists between the ionized
fentanyl (F+
), the free base (F), and the fentanyl molecules bound to
macromolecules (Fb
). The concentration of ionized fentanyl within the
interstitial fluid (encircled F+
) determines the pharmacologic effect,
because the opioid receptors are located at the cell surface. Fentanyl as free base
readily penetrates into the cells and becomes bound to cytomembranes, lysosomes,
and other structures (thick arrows) and thus accumulates
in the cell. A small decrease of the extracellular pH will shift the equilibrium
between F and F+
toward higher F+
concentrations and will induce
a marked release of fentanyl from the cellular compartment as a result of the decrease
of the concentration of F in the interstitium. An increase of the extracellular
H+
concentration by pH 0.2 might roughly double the concentration of ionized
fentanyl within the interstitial fluid and accordingly enhance its pharmacologic
effect. (From Lullmann H, Martins BS, Peters T: pH-dependent accumulation
of fentanyl, lofentanil and alfentanil by beating guinea pig atria. Br J Anaesth
57:1012–1017, 1985.)
the decline of opioid concentration, particularly in the early period after injection.
Opioid uptake by the lung is a significant implication of opioid
pharmacokinetics. The time necessary to reach peak concentration of an opioid is
influenced by the percentage of pulmonary uptake. A very substantial proportion
of the initial dose of a highly lipophilic opioid such as fentanyl is taken up by
the lung (75%) and subsequently is rapidly released.[296]