|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Patients requiring cardiac surgery may have varying degrees of chronic preexisting myocardial dysfunction, or myocardial dysfunction may develop acutely after aortic cross-clamping and anoxic arrest for any of several reasons (see Table 50-17 ). Moreover, many cases of perioperative myocardial dysfunction represent acute perioperative exacerbations of preexisting chronic dysfunction. Surgery in patients with chronic heart failure is performed with the objective of either addressing the underlying cause or managing end-stage heart failure per se. In addition to the acute, chronic, and acute-on-chronic circumstances outlined, myocardial dysfunction may involve the left or the right ventricle, or both, and may be manifested primarily as systolic or diastolic dysfunction, or both. The challenge for the perioperative physician is to identify the predominant problem and manage the patient accordingly ( Table 50-18 ), which requires an understanding of the underlying mechanisms
| Exacerbation of preexisting dysfunction and relative intolerance of anoxic arrest |
| Inadequate myocardial protection (role of underlying coronary anatomy, route of cardioplegia administration, type of cardioplegia) |
| Ischemia/infarction |
| Vessel spasm (native coronaries, internal mammary artery) |
| Emboli (air, particulate) |
| Technical graft anastomotic tissues |
| Kink/clotting of grafts, native vessels |
| Nongraftable vessels |
| Reperfusion injury |
| Unmasked ventricular dysfunction (mitral valve replacement/repair with mitral regurgitation) |
| Uncorrected lesions |
| Hypertrophic cardiomyopathy |
| Valve gradients |
| Shunts |
Enormous advances have been made over the last 20 years in our
understanding of the mechanistic changes that occur in heart failure.[286]
Although the inciting event in heart failure may be cardiac (e.g., myocardial infarction,
valve disease) or extracardiac (chronic ventricular overload) ( Fig.
50-44
), it is the reflex responses to this inciting event that are mechanistically
responsible for the seminal functional and structural features of heart failure.
Reflex neurohumoral activation of the autonomic nervous system, the renin-angiotensin
system, and arginine vasopressin resulting in what was believed to be an "adaptive"
response promoting adequate blood pressure and organ perfusion is long outdated.
It is now clear that multiple mechanisms with varying degrees of redundancy and
crosstalk are activated in heart failure. Moreover, many of these pathways result
in "maladaptive" changes in the heart, including trophic changes ( Fig.
50-45
). Progressive heart failure should be viewed as resulting from changes
in multiple interrelated mechanisms
| Identify the treatment course (if possible) |
| Optimize forward flow (heart rate, rhythm [± pacing]), manipulate loading conditions and inotropic and lusitropic conditions (glyceryl trinitrate, sodium nitroprusside, epinephrine, dobutamine) |
| Treat acidosis |
| If right ventricular failure, optimize specific afterload determinants, i.e., PO2 , PCO2 , pH, airway/intrathoracic pressure, nitrates, inhaled nitric oxide |
| Mechanical circulatory support systems |
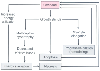 Figure 50-44
Some of the vicious cycles that operate in an overloaded
heart. Overload both increases energy utilization and stimulates growth (dark
arrows). The former contributes directly to a state of energy starvation,
which is made worse by several consequences of maladaptive hypertrophy that decrease
the energy supply. The latter includes myocyte elongation, which causes remodeling,
a progressive dilatation that increases wall tension and thereby increases the overload.
Growth stimuli also promote apoptosis, which by decreasing the number of viable
cardiac myocytes, increases the load on those that survive. Hypertrophy also causes
architectural changes that reduce the energy supply to working cardiac myocytes.
(Redrawn from Katz LN: Maladaptive hypertrophy and the cardiomyopathy of
overload: Familial cardiomyopathies. In Katz AM
[ed]: Heart Failure: Pathophysiology, Molecular Biology, and Clinical Management.
Philadelphia, Lippincott Williams & Wilkins, 2000, pp 277–307.)
Figure 50-44
Some of the vicious cycles that operate in an overloaded
heart. Overload both increases energy utilization and stimulates growth (dark
arrows). The former contributes directly to a state of energy starvation,
which is made worse by several consequences of maladaptive hypertrophy that decrease
the energy supply. The latter includes myocyte elongation, which causes remodeling,
a progressive dilatation that increases wall tension and thereby increases the overload.
Growth stimuli also promote apoptosis, which by decreasing the number of viable
cardiac myocytes, increases the load on those that survive. Hypertrophy also causes
architectural changes that reduce the energy supply to working cardiac myocytes.
(Redrawn from Katz LN: Maladaptive hypertrophy and the cardiomyopathy of
overload: Familial cardiomyopathies. In Katz AM
[ed]: Heart Failure: Pathophysiology, Molecular Biology, and Clinical Management.
Philadelphia, Lippincott Williams & Wilkins, 2000, pp 277–307.)
The essential role of diastolic function in myocardial performance, that is, the ability of the heart to fill, is clearly recognized. Dysfunction implies an abnormal index of diastolic function, whereas failure implies an associated clinical syndrome. The mechanisms underlying diastolic performance and quantitative techniques to measure diastolic performance are increasingly being understood. With this has come an increased understanding of when diastolic dysfunction may undermine cardiac performance, the underlying mechanisms, and potential therapies. The clinical focus on systolic function (i.e., the ability of the heart to empty), especially perioperatively, reflects our ability to quantify it (at least globally with echocardiography and cardiac output measurements), our understanding of the underlying mechanisms, and our ability to favorably examine these mechanisms pharmacologically ( Fig. 50-46 ).
This situation contrasts with diastolic function, where the best available clinical measures involve Doppler interrogation of mitral valve flow, pulmonary vein velocities, and Doppler tissue techniques. Laboratory techniques that are used to measure diastolic function include tau (τ), the time that it takes for left ventricular pressure to decrease by two thirds during isovolumic relaxation,
 Figure 50-45
A, Original neurohumoral
model of congestive heart failure (CHF). B, Current
neurohumoral paradigm of CHF, with an emphasis on the central role of the heart.
(A, Redrawn from Francis GS, Goldsmith SR,
Levine TB, et al: The neurohumoral axis in congestive heart failure. Ann Intern
Med 101:370–377, 1984. B, Redrawn from McMurray
J, Pfeffer MA: New therapeutic options in congestive heart failure: Part I. Circulation
105:2099–2106, 2002.)
Figure 50-45
A, Original neurohumoral
model of congestive heart failure (CHF). B, Current
neurohumoral paradigm of CHF, with an emphasis on the central role of the heart.
(A, Redrawn from Francis GS, Goldsmith SR,
Levine TB, et al: The neurohumoral axis in congestive heart failure. Ann Intern
Med 101:370–377, 1984. B, Redrawn from McMurray
J, Pfeffer MA: New therapeutic options in congestive heart failure: Part I. Circulation
105:2099–2106, 2002.)
The prevalence and significance of diastolic dysfunction in the
general population are unknown. However, diastolic dysfunction was observed in 50%
of patients older than 70 years who underwent diagnostic screening.[289]
[290]
In patients with diastolic dysfunction and
preserved systolic function, the annual mortality is 5% to 8%.[291]
[292]
The prevalence of perioperative diastolic
dysfunction (either new onset or exacerbation of preexisting diastolic
| Extramyocardial |
| Hemodynamic load: early diastolic load, afterload |
| Heterogeneity |
| Pericardium |
| Myocardial |
| Cardiomyocyte |
| Calcium hemostasis |
| Calcium concentration |
| Sarcolemmal and SR calcium transport function |
| Modifying proteins (phospholamban, calmodulin, calsequestrin) |
| Myofilaments |
| Troponin C-calcium binding |
| Troponin I phosphorylation |
| Myofilament calcium sensitivity |
| α/β-Myosin heavy chain-ATPase ratio |
| Energetics |
| ADP/ATP ratio |
| ADP and Pi concentration |
| Cytoskeleton |
| Microtubules |
| Intermediate filaments (desmin) |
| Microfilaments (actin) |
| Endosarcomeric skeleton (titin, nebulin) |
| Extracellular matrix |
| Fibrillar collagen |
| Basement membrane proteins |
| Proteoglycans |
| MMP/TIMP |
| Neurohormonal activation |
| Renin-angiotensin-aldosterone |
| Sympathetic nervous system |
| Endothelin |
| Nitric oxide |
| Natriuretic peptides |
| ADP, adenosine diphosphate; ATP, adenosine triphosphate; MMP, matrix metalloproteinase; Pi , inorganic phosphate; SR, sarcoplasmic reticulum; TIMP, tissue inhibitor of metalloproteinases. |
| From Zile MR, Brutsaert DL: New concepts in diastolic dysfunction and diastolic heart failure: Part II: Causal mechanisms and treatment. Circulation 105:1503–1508, 2002. |
Treatment of diastolic heart failure presumes that we can diagnose the entity and measure the response to an intervention. Such is not the case at this time. Conceptually, however, treatments should be directed at alleviating symptoms (e.g., pulmonary congestion with nitrates), treating the cause of the diastolic failure (e.g., ischemia), and interrogating the mechanisms activated by the disease process (e.g., the renin-angiotensin system). Many of the drugs used to treat diastolic and systolic failure (see Fig. 50-46 ) are the same (e.g., β-blockers, nitrates). However, it is important to recognize that this is not always the case, and even when the same drug is used, the dosing regimen may be importantly and subtly different for systolic and diastolic failure. For example, the calcium antagonists now used for diastolic failure are contraindicated in systolic failure. β-Blockers can be used acutely in diastolic failure, where the salutary effects are related to a decrease in heart rate and an increase in the diastolic
 Figure 50-46
A and B,
Signal systems involved in positive inotropic and lusitropic (enhanced relaxation)
effects of β-adrenergic stimulation. When the β-adrenergic agonist interacts
with the β-receptor, a series of G protein-mediated changes lead to activation
of adenylate cyclase and the formation of cyclic adenosine monophosphate (cAMP).
The latter acts via protein kinase A to stimulate metabolism (left)
and phosphorylate the calcium channel protein. The result is an enhanced probability
of the calcium channel being open, thereby increasing the inward movement of Ca2+
ions through the sarcolemma (SL) of the T tubule. These Ca2+
ions release
more calcium from the sarcoplasmic reticulum (SR) to increase cytosolic calcium and
activate troponin C. Calcium ions also increase the rate of breakdown of adenosine
triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate (Pi
).
Enhanced myosin ATPase activity explains the increased rate of contraction, with
increased activation of troponin C explaining the development of increased peak force.
An increased rate of relaxation is explained by the fact that cAMP also activates
the protein phospholamban (PL), which is situated on the membrane of the SR and controls
the rate of uptake of calcium into the SR. The latter effect explains enhanced relaxation
(lusitropic effect). TnI, troponin I. (From Opie LG: Receptors and signal
transduction. In Opie LH [ed]: The Heart, Physiology
from Cell to Circulation, 4th ed. Philadelphia, Lippincott Raven, 1997, pp 173–207.)
Figure 50-46
A and B,
Signal systems involved in positive inotropic and lusitropic (enhanced relaxation)
effects of β-adrenergic stimulation. When the β-adrenergic agonist interacts
with the β-receptor, a series of G protein-mediated changes lead to activation
of adenylate cyclase and the formation of cyclic adenosine monophosphate (cAMP).
The latter acts via protein kinase A to stimulate metabolism (left)
and phosphorylate the calcium channel protein. The result is an enhanced probability
of the calcium channel being open, thereby increasing the inward movement of Ca2+
ions through the sarcolemma (SL) of the T tubule. These Ca2+
ions release
more calcium from the sarcoplasmic reticulum (SR) to increase cytosolic calcium and
activate troponin C. Calcium ions also increase the rate of breakdown of adenosine
triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate (Pi
).
Enhanced myosin ATPase activity explains the increased rate of contraction, with
increased activation of troponin C explaining the development of increased peak force.
An increased rate of relaxation is explained by the fact that cAMP also activates
the protein phospholamban (PL), which is situated on the membrane of the SR and controls
the rate of uptake of calcium into the SR. The latter effect explains enhanced relaxation
(lusitropic effect). TnI, troponin I. (From Opie LG: Receptors and signal
transduction. In Opie LH [ed]: The Heart, Physiology
from Cell to Circulation, 4th ed. Philadelphia, Lippincott Raven, 1997, pp 173–207.)
Both systolic and diastolic heart failure involve multiple mechanisms. It is unlikely that modulation of one or even a few of these pathways is likely to have a significant impact on the natural history of this disease. Effective management of heart failure will require modulation of multiple pathways. Currently used therapies involving the renin-angiotensin system include ACE inhibitors, angiotensin receptor blockers, and aldosterone antagonists. β-Blockers, which affect the autonomic system, are also in standard use. Natriuretic peptide analogs such as nesiritide, the human analog of type β natriuretic peptide,
The degrees of heart failure and the management responses have been cunningly illustrated in a cartoon by Katz ( Fig. 50-47 ).[286] In the intraoperative setting, we use at least four of these approaches. In acute systolic heart failure we both beat the horse (inotropes) and unload the wagon (pharmacologically induced afterload reduction, [IAPB]). We probably would slow the horse (β-blockers) in diastolic failure if we had a reliable means of diagnosing it and if it occurred without systolic failure. For patients with chronic end-stage heart failure, we either replace the horse (transplantation) or get a tractor (mechanical circulatory support system), both fraught with problems and less than ideal. Although some causes of acute failure can be addressed in the operating room (e.g., ischemia-induced failure reversed by revascularization), the real societal problem is the increasing epidemic of chronic CHF. The fundamental solution to this problem is to "heal the horse," a solution that will probably emerge over the next decades as we increase our mechanistic understanding of heart failure, and it is highly unlikely to involve the operative setting.
Post-CPB ventricular dysfunction may represent an exacerbation of existing ventricular dysfunction or may develop in the setting of reasonable preoperative function. The more common causes are listed in Table 50-17 .
 Figure 50-47
View of the failing heart as a sick, tired horse pulling
a wagon up a steep hill. Although application of a whip (inotropes) encourages the
horse to move faster, such treatment can kill the animal. Unloading the wagon (vasodilators)
would seem to be advantageous, but in heart failure, this approach can harm the horse
by activating harmful neurohumoral responses. Slowing the horse (β-adrenergic
blockers) while delaying the journey can be beneficial, especially if this also helps
heal the horse. Replacing the horse (cardiac transplantation) is useful as long
as enough spare horses are available, and getting a tractor is a solution only if
reliable machines are available. The ideal solution, of course, is to learn what
ails the animal and to use this information to heal the horse. (From Silber
EN, Katz LN: Therapeutic strategies for managing heart failure. In
Katz AM [ed]: Heart Failure: Pathophysiology, Molecular Biology and Clinical Management.
Philadelphia, Lippincott Williams & Wilkins, 2000, pp 309–339.)
Figure 50-47
View of the failing heart as a sick, tired horse pulling
a wagon up a steep hill. Although application of a whip (inotropes) encourages the
horse to move faster, such treatment can kill the animal. Unloading the wagon (vasodilators)
would seem to be advantageous, but in heart failure, this approach can harm the horse
by activating harmful neurohumoral responses. Slowing the horse (β-adrenergic
blockers) while delaying the journey can be beneficial, especially if this also helps
heal the horse. Replacing the horse (cardiac transplantation) is useful as long
as enough spare horses are available, and getting a tractor is a solution only if
reliable machines are available. The ideal solution, of course, is to learn what
ails the animal and to use this information to heal the horse. (From Silber
EN, Katz LN: Therapeutic strategies for managing heart failure. In
Katz AM [ed]: Heart Failure: Pathophysiology, Molecular Biology and Clinical Management.
Philadelphia, Lippincott Williams & Wilkins, 2000, pp 309–339.)
Effective management of post-CPB ventricular dysfunction extends beyond optimizing the heart rate, rhythm, loading conditions, and augmenting inotropy and lusitropy; it also involves identification of the cause of failure, the primary ventricle involved, and treatment of any consequences of inadequate organ perfusion (e.g., acidosis) (see Table 50-18 ). The importance of diastolic failure after CPB is increasingly being recognized and was discussed earlier. The causes of right ventricular dysfunction are outlined in Table 50-20 . Importantly, right ventricular ischemia is a major cause of right ventricular dysfunction.[300] Note also that in the clinical setting, one cause of right ventricular dysfunction might predominate, but many other factors may also contribute. For example, borderline ischemia with right ventricular dysfunction may be unmasked by an increase in right ventricular afterload, which itself can be caused by a myriad of conditions. The seminal clinical features of right ventricular dysfunction are right atrial pressure greater than 20 mm Hg, left atrial pressure less than 10 mm Hg, and a cardiac index less than 1.8 to 2 L/min/m2 . Cardiac output decreases as central venous pressure increases. Depending on the circumstances, pulmonary artery pressure may be elevated but could, in extreme circumstances, be lower than what might be anticipated simply because of right ventricular failure. The right ventricle is dilated, hypocontractile, and volume overloaded. In addition to these latter features being demonstrable on echocardiography, one may observe bowing of the interventricular septum into the left ventricle and
| Right ventricular ischemia |
| Pulmonary hypertension and elevated pulmonary vascular resistance |
| Pulmonary arterial hypertension |
| Primary pulmonary hypertension |
| Sporadic |
| Familial |
| Related to |
| Collagen vascular disease |
| Congenital systemic-to-pulmonary shunts |
| Portal hypertension |
| Human immunodeficiency virus infection |
| Drugs/toxins |
| Anorexigens |
| Other |
| Persistent pulmonary hypertension of the newborn |
| Pulmonary venous hypertension |
| Pulmonary hypertension associated with disorders of the respiratory system and/or hypoxemia (e.g., chronic obstructive pulmonary disease, interstitial lung disease, sleep disordered breathing, alveolar hypoventilation disorders, pulmonary hypertension caused by chronic thrombotic and/or embolic disease) |
| Altered interventricular dependence (e.g., after placement of a left ventricular assist device)[1] |
| From Chen JM, Levin HR, Rose EA, et al: Experience with right ventricular assist devices for perioperative right-sided circulatory failure. Ann Thorac Surg 61:305–310, 1996. |
In its broadest definition, mechanical assist devices include (1) CPB, (2) IABP counterpulsation, and (3) mechanical circulatory support (MCS) systems. CPB has already been discussed.
IABP was first introduced clinically in 1968.[301]
The central feature of IABP counterpulsation involves the alternating inflation
(during diastole) and deflation (during
| Treat course |
| Optimize preload (central venous pressure: 12–15 mm Hg) |
| Establish sinus rhythm with a reasonable rate |
| Inotropic support |
| Afterload reduction |
| Nonspecific: optimize blood gases, pH, and airway pressures |
| Specific: phosphodiesterase inhibitors, nitrosovasodilators, nitric oxide |
| Mechanical assist device |
The two major indications for IABP placement are myocardial ischemia that is intractable to maximal medical therapy and left ventricular dysfunction inadequately managed with inotropic therapy. The latter is a common indication for intraoperative, post-CPB placement, whereas the former indication is more frequently encountered in the coronary care unit, where it is used as a stabilizing measure before definitive intervention (e.g., angioplasty, stent placement, surgery). Properly timed counterpulsation of an appropriately placed IABP has several beneficial hemodynamic effects, including (1) increasing diastolic blood pressure; (2) decreasing systolic blood pressure; (3) decreasing left ventricular systolic work, tension, and myocardial oxygen consumption; and (4) decreasing afterload. The net effect is to favorably influence the determinants of myocardial oxygen supply and demand and augment left ventricular output and forward flow.
IABP counterpulsation may also have favorable effects on right ventricular function. Although the mechanisms are complex, they probably include accentuation of right ventricular myocardial blood flow, unloading of the left ventricle with decreases in left atrial and pulmonary vascular pressures and right ventricular afterload, and improvements in right ventricular mechanical function secondary to changes in left ventricular performance and ventricular interdependence. The efficacy of IABP counterpulsation is critically dependent on proper placement in the aorta and proper timing of inflation and deflation. The IABP should be positioned as close as possible to the heart, but distal to the great vessels, and should be inflated in synchrony with the dicrotic notch on an arterial pressure trace. Optimal timing can be determined by using the patient's arterial blood pressure tracing or the patient's ECG, which can be monitored directly by the IABP console or indirectly by standard operating room monitors.
IABP counterpulsation is contraindicated in patients with aortic incompetence and arterial dissection and is relatively contraindicated in patients with severe atherosclerosis. Complications include thromboembolic phenomena, distal limb ischemia (to which thromboembolism and mechanical obstruction of the femoral artery may contribute), thrombocytopenia, gas emboli (rupture of the balloon), and infection.
The influence of several factors is likely to increase the use of these systems in the future. The indication for
The overall goal of MCS is not only to improve the patient's quality of life but also to extend that life. MCS can be used as a bridge to recovery, as a bridge to transplantation, and as long-term replacement or destination therapy. A total artificial heart has not yet been developed to the point that it is approved for general use. The bridge-to-recovery approach is used in patients who (1) sustain postsurgical myocardial dysfunction not adequately supported by inotropes and IABP counterpulsation, (2) suffer acute heart failure (e.g., secondary to myocarditis), and (3) have post-myocardial infarction cardiogenic shock but retain the potential for a meaningful recovery. Some investigators exclude patients with creatine phosphokinase levels over 10,000 and troponin levels over 300.[305] The bridge-to-transplantation strategy is used in patients with CHF. Note that the bridge-to-transplantation category is not absolutely exclusive of the bridge-to-recovery category. It is now well recognized that the improved hemodynamic conditions and neurohumoral milieu that occurs with MCS in patients hitherto severely compromised hemodynamically can result in end-organ remodeling to an extent that may preclude the requirement of proceeding to transplantation.[306] [307] [308] [309] [310]
A number of MCS systems are currently available,[302] [305] [311] and they vary from each other in several important features, including (1) the mechanism used to propel blood (the so-called actuation mechanism), (2) size, (3) surface material in contact with blood and its influence on anticoagulant use, and (4) the ability to support the right as well as the left side of the heart versus the left side only ( Table 50-22 ).
The Abiomed BVS 5000 system is an external mechanical system that
can be used for univentricular or biventricular support. Bleeding and thromboembolic
and infectious complications limit its use to less than 14 days. Thus,
|
|
Ventricular Support | Surface Lining | Long-term Anticoagulation | Influence of Patient Size | Duration of Use |
|---|---|---|---|---|---|
| Abiomed BVS 5000 | Left and right | Smooth | Heparin/warfarin | — | <14 days |
| Heartmate | Left | Textured | Antiplatelet agents | Difficult to insert in small patients | Long term |
| Novocor | Left | Smooth | Heparin/warfarin | — | Long term |
| Thorotec | Left and right | Smooth | Heparin/warfarin | — | Long term |
The Heartmate and Novocor devices are both used for long-term support of the left ventricle. They differ from each other in certain important features. The Heartmate device is lined by a "textured" surface, which promotes the deposition of fibrin and circulating cells. This feature allows patients to be managed long-term with antiplatelet agents only and decreases hemorrhagic complications. Infectious complications are also thought to be decreased. The Novocor device requires systemic anticoagulation with warfarin (Coumadin). The Heartmate device can propel blood by using either a pneumatic system or an electrically driven motor system. In the Novocor system, blood propulsion depends on the rhythmic compression of a polyurethane blood-filled bag by two opposing plates. Both systems require CPB for placement. The cannula connections are similar in both systems, with blood drained from the apex of the left ventricle to the pumping mechanism, which is implanted either intraperitoneally or preperitoneally below the left costal margin. The blood is returned via a cannula inserted into the ascending aorta. In both systems, one-way valves ensure forward flow. The pumping mechanism is connected to an external, portable console, which provides a power source and contains the control unit.
The Thoratec device is pneumatically driven and can be configured for the left, right, or both ventricles. On the left side, the outflow cannula can be placed in either the left ventricle or the left atrium with the inflow cannula in the ascending aorta. On the right side, the outflow and inflow cannulas are placed in the right atrium and pulmonary artery, respectively. Cannula placement does not necessarily require CPB. The cannulas are connected to externally placed pumps, which are themselves connected to an external console. The external location of the pump mechanism may confer certain advantages. It may be more easily used in smaller patients, and the pump can be changed if it malfunctions or if infection or thrombosis occurs. The smooth polyurethane surfaces dictate that systemic anticoagulation be used.
At the time of writing, the Cardio-West Total Artificial Heart is approved for use under an FDA investigational device exemption. This pneumatically driven system is placed in the native heart's position, and dual drivelines are externalized to connect with an external console.
The Heartmate II, MicroMed DeBakey VAD, and Jarvik 2000 systems are also being evaluated regarding their suitability to function as MCS systems. The Jarvik 2000 is an axial (i.e., continuous) flow device inserted within the left ventricle, with blood being pumped into the ascending aorta. It provides partial left ventricular support and may have potential in suitably selected patients.[312] [313]
|
|
|
|
|
|
|
|
|
|
|
|
|