|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The cholinergic drugs act by mimicking, amplifying, or inhibiting the effects of acetylcholine. Cholinergic drugs do not behave exactly as does acetylcholine; their drug action is more specific, affecting fewer sites than acetylcholine, and their duration of action is generally longer than that of acetylcholine.
Unlike adrenergic pharmacology, in which the clinician can select from a wide choice of drugs, there is a relative paucity of drugs that influence parasympathetic function. In general, drugs that affect the parasympathetic system act in one of four ways:
There are no effective clinically used drugs that act through mechanisms affecting synthesis of acetylcholine (by inhibiting choline acetyltransferase) or by causing indirect release of acetylcholine (because tyramine, ephedrine, and amphetamine may release norepinephrine). Hemicholinium, which interferes with choline uptake and could deplete acetylcholine stores, is not used clinically. Adenosine may inhibit the release of acetylcholine by decreasing the affinity of binding sites for calcium ions; aminoglycoside antibiotics may compete with calcium for membrane calcium channels, as does magnesium ion. Exocytotic release of acetylcholine is inhibited by botulinum toxin; this toxin is sometimes given by local injection to treat strabismus and blepharospasm. Botulinum toxin has also been used for trigger point injections in pain clinics and to treat age lines. The popularity of off-label uses of this toxin increases the chances of botulism poisoning. In a full-blown botulism-poisoning syndrome, fatalities may result from muscle weakness and respiratory failure.
Cholinergic agonists have limited therapeutic use because of their detrimental effects. As a result of its diffuse, nonselective actions and rapid hydrolysis by acetylcholinesterase and butyrylcholinesterase, acetylcholine
Cholinergic agonists in clinical use have been derived from acetylcholine, but they resist hydrolysis by cholinesterase, permitting a useful duration of action. The different systemic effects of the cholinergic agonists are more quantitative than qualitative, but some limited organ selectivity is useful therapeutically, as is seen with the synthetic choline esters bethanechol and carbachol. Methacholine and bethanechol are primarily muscarinic agonists; carbachol has significant nicotinic and muscarinic effects. The simple maneuver of adding a methyl group to the β-position of the choline in acetylcholine produces methacholine, which is almost purely muscarinic and is almost totally resistant to hydrolysis by either of the cholinesterases. An intravenous infusion of methacholine causes hypotension and bradycardia; a small subcutaneous dose causes transient hypotension with a reflex increase in heart rate. The sole current use of methacholine (Provocholine) is as a provocative agent in diagnosing hyperreactive airways, making positive use of the deleterious bronchoconstrictive effect of muscarinic agonists. It is administered only by inhalation; serious adverse effects include gastrointestinal symptoms, chest pain, hypotension, loss of consciousness, and complete heart block when the drug is given orally or parenterally. Excessive bronchoconstrictive response should be treated by an inhaled β-agonist; coexisting β-blockade is considered a contraindication to the use of methacholine.
The carbamate derivative of methacholine, bethanecol (Urecholine), is occasionally used postoperatively to reinstitute peristaltic activity in the gut or to force the extrusion of urine from an atonic bladder. It is administered subcutaneously or orally to avoid effects in other organ systems.
Carbachol is used topically or intraocularly to constrict the pupil for long-term treatment of wide-angle glaucoma. When used topically, it is often better tolerated than the ophthalmic anticholinesterase agents, and it may be effective in patients resistant to pilocarpine and physostigmine. The rapid pupillary constriction is caused by the combination of ganglionic block and muscarinic effects. Another natural alkaloid, pilocarpine, was used to treat glaucoma until the advent of more modern drugs.
Muscarinic antagonists are the active ingredients in some common
plants used since antiquity for medicinal and
| Drug | Duration | Central Nervous System * | Antisialagogue | Heart Rate |
|---|---|---|---|---|
| Atropine | Short | Stimulation | + | ++ |
| Glycopyrrolate | Long | 0 | ++ | + |
| Scopolamine | Short | Sedation | ++ | 0/+ |
| 0, no effect; +, mild effect, ++, moderate effect. | ||||
Muscarinic antagonists compete with neurally released acetylcholine for access to muscarinic cholinoceptors and block its effects. They also antagonize the actions of muscarinic agonists at noninnervated, muscarinic cholinoceptors. Presynaptic muscarinic receptors on the adrenergic nerve terminal may inhibit norepinephrine release, and muscarinic antagonists may enhance sympathetic activity. With the exception of quaternary ammonium compounds that do not readily cross the blood-brain barrier and have few CNS actions, there is no significant specificity of action among these drugs; they block all muscarinic effects with equal efficacy, although some quantitative differences in effect may be seen ( Table 16-12 ). Research has revealed several subtypes of muscarinic receptors, and agonists and antagonists have been synthesized that bind preferentially to one or another. None of these selective agents is yet available commercially, but with this rapidly developing area of research, there will soon be drugs that selectively act on one site or another, such as the heart, bronchial system, smooth muscle, gastric mucosa, or specific areas of the CNS.
Historically, these drugs were used in peptic ulcer disease, various forms of spastic bowel syndrome, upper respiratory illness, and asthma. However, with the availability of the specific histamine (H2 )-blocking drugs such as cimetidine for peptic ulcer disease, these uses have markedly decreased. Atropine, once used to treat bronchospasm, was displaced with the introduction of β2 -agonist drugs that did not dry secretions or diminish ciliary motility. Topical use of atropine analogs in ophthalmologic practice to dilate the pupil is still common.
The addition of a muscarinic anticholinergic drug to anesthetic premedication to decrease secretions and to prevent harmful vagal reflexes was mandatory in the era of ether anesthesia, but it is no longer necessary with modern inhaled agents. Scopolamine combined with an opiate, usually morphine, is still used by cardiac anesthesiologists to sedate a patient while minimizing cardiorespiratory effects. Routine preoperative use of these drugs as antisialagogues continues in some pediatric and otorhinolaryngologic cases or when fiberoptic intubation is planned.
Atropine has a tertiary structure that easily crosses the blood-brain barrier ( Fig. 16-22 ). CNS effects have been seen with the relatively large doses (1 to 2 mg) given to block the muscarinic adverse effects of the anticholinesterase drugs
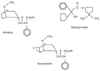 Figure 16-22
Structural formulas of the clinically useful antimuscarinic
drugs.
Figure 16-22
Structural formulas of the clinically useful antimuscarinic
drugs.
Scopolamine, which resembles the others of this class in peripheral actions, has pronounced CNS effects. It is the active ingredient in most over-the-counter preparations sold as soporifics, and it is effective in preventing motion sickness. The patch preparation of scopolamine can be used prophylactically for motion sickness and for postoperative nausea and vomiting, but like oral and parenteral forms, it may be associated with eye, bladder, skin, and psychological adverse effects.[436] [437]
The development of ipratropium (Atrovent) reestablished antimuscarinic drugs for the treatment of asthma and bronchospastic disorders.[438] Although ipratropium is structurally similar to atropine and has essentially the same effects when administered parenterally, an important difference is that ipratropium is a quaternary ammonium compound. It is very poorly absorbed when inhaled and has few extrapulmonary effects, even in extremely large doses by this route. Ninety percent of inhaled drug is swallowed, but only 1% of the total dose is absorbed systemically.
When administered to normal volunteers, ipratropium provides almost complete protection against bronchospasm induced by a variety of provocative agents. However, in asthmatic patients, results vary. The bronchospastic effects of some agents, such as methacholine or sulfur dioxide, are completely blocked, whereas there is little effect on leukotriene-induced bronchoconstriction. The onset of bronchodilation is slow, and the maximum effect is less than that seen with β-agonists. Unlike atropine, ipratropium has no negative effect on ciliary clearance. In general, therapeutic effect from antimuscarinics, including ipratropium, is greater in patients with chronic obstructive pulmonary disease than in asthmatic patients.[438] [439] Ipratropium is supplied as a metered-dose inhaler dispensing 18 µg per puff. Dosage is two puffs orally four times each day. Maximum bronchodilation occurs in 30 to 90 minutes and may last 4 hours.[439]
The toxic effects of the muscarinic antagonists come from the blockade of muscarinic cholinoceptors in the periphery and the CNS. The peripheral effects (e.g., dry mouth) may be irritating but are not life threatening in healthy adults. However, children depend more than adults on sweating for thermoregulation and easily become dangerously hyperthermic. Moreover, older individuals may not be able to tolerate the cardiac, ocular, or urinary effects of muscarinic blockade.
The CNS effects are the usual cause of death or injury. Increasing doses of atropine or scopolamine cause greater distortions of mentation, progressing from thought disorders to hallucinations, delusions, delirium, and severe psychoses. These effects are reversible, but the mental dysfunction can persist for weeks. Left alone, the intoxicated individual will die of starvation, dehydration, or trauma. Those who have received more than 500 mg of atropine, which is more than 1000 times the usual dose, and have been disabled for weeks have recovered fully.
Small doses of atropine (0.05 mg) can evoke bradycardia, a finding that has led some clinicians to increase the dose in children. It was thought that a CNS effect of
Atropine and scopolamine toxicity have been treated for decades by the use of the naturally occurring alkaloid physostigmine (Antilirium), which is an anticholinesterase that penetrates the blood-brain barrier.[441] The use of this drug in doses of 1 to 2 mg given intravenously to treat the postoperative CNS effects of intravenous atropine or scopolamine has been successful. Physostigmine may also reverse the CNS effects of other compounds with anticholinergic activity, including the tricyclic antidepressants, several major tranquilizers, and antihistamine drugs.[442] Physostigmine also may antagonize the sedative effects of the benzodiazepines, but a specific benzodiazepine antagonist, flumazenil (Romazicon), has supplanted physostigmine for this use.[443] [444] Physostigmine must be administered with care because of its potentially lethal nicothinic effects, which are not prevented by the muscarinic antagonists, and because its half-life rarely matches that of the intoxicant.
Anticholinesterase drugs are a common means of producing sustained, systemic cholinergic agonism. These drugs are used to reverse neuromuscular blockade, to treat myasthenia gravis, and to treat certain tachyarrhythmias.
The first anticholinesterase agent available was physostigmine. There are three chemical classes of compounds used as cholinesterase inhibitors: carbamates, organophosphates, and quaternary ammonium alcohols. Neostigmine was first used as a gastrointestinal tract stimulant and later as a treatment for myasthenia gravis.
Physostigmine, neostigmine, and pyridostigmine are carbamates, whereas edrophonium is a quaternary ammonium alcohol. The cholinesterase enzyme is inhibited so long as the esteratic site is bound to an acetate, carbamate, or phosphate. Carbamate and phosphate bonds are much more resistant to attack by hydroxyl groups than are acetate bonds. The acetylated form lasts for only microseconds, whereas the carbamylated form lasts for 15 to 20 minutes. Organophosphates include diisopropyl fluorophosphate, parathion, Malathion, soman, sarin, VX, and a variety of other compounds used as insecticides. Although the toxicity of the organophosphate insecticides is primarily related to their anticholinesterase activity, the mechanism of this effect is different from the clinically used anticholinesterase drugs. The organophosphates produce an irreversible enzyme inhibition and have CNS effects. [445] Consequently, treatment of organophosphate insecticide poisoning relies on chemical compounds capable of displacing the insecticides from the enzyme to reactivate the cholinesterase activity. The best documented of these chemicals is pralidoxime (2-PAM). Physostigmine and most of the organophosphates are not quaternary ammonium compounds and have major effects on cholinergic functions in the CNS.
Edrophonium is unique in that it lacks an acetate, carbamate, or phosphate group. It acts because the positive charge of the nitrogen is attracted strongly by the anionic site and physically blocks the esteratic site. The edrophonium molecule is postulated to be held in place only by an ionic bond. The duration of inhibition provided by each molecule is short (milliseconds), but because they are not changed in the reaction, the molecules can hop onto and off the enzyme repeatedly to render the enzyme unavailable to acetylcholine.
Aside from reversal of neuromuscular blockade, there are few other therapeutic uses of these compounds. Because these compounds can increase the effect and duration of neurally released acetylcholine, they are useful in situations in which such release is deficient, such as myasthenia gravis. Anticholinesterase drugs are occasionally used to stimulate intestinal function and used topically in the eye as a miotic. An irreversible organophosphate anticholinesterase that is used clinically is echothiophate iodide (Phospholine), which is available as topical drops for the treatment of glaucoma. Its major advantage over other topical agents is its prolonged duration of action. Because this chemical also inactivates plasma cholinesterase, it may prolong the action of succinylcholine. Although prudence dictates discontinuation of echothiophate for 1 week before surgery, there are numerous case reports of successful anesthesia performed without discontinuation of echothiophate under emergency conditions.
|
|
|
|
|
|
|
|
|
|
|
|
|