|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In contrast to the diffuse discharge of the sympathetic nervous system that constitutes the fight or flight
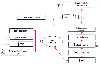 Figure 16-7
The interactions of the renin-angiotensin-aldosterone
and sympathetic nervous systems in maintaining blood pressure and volume. Al, angiotensin
I; All, angiotensin II; CE, converting enzyme; NE, norepinephrine; RBF, renal blood
flow; +, stimulating effects; -, inhibiting effects.
Figure 16-7
The interactions of the renin-angiotensin-aldosterone
and sympathetic nervous systems in maintaining blood pressure and volume. Al, angiotensin
I; All, angiotensin II; CE, converting enzyme; NE, norepinephrine; RBF, renal blood
flow; +, stimulating effects; -, inhibiting effects.
Acetylcholine release is the hallmark of parasympathetic activation. The actions of acetylcholine are almost diametrically opposed to those of norepinephrine and epinephrine. In general, the muscarinic effects of acetylcholine are qualitatively the same as the effects of vagal stimulation. Acetylcholine is the only endogenous compound that causes simultaneous bradycardia and hypotension.
The dose of acetylcholine determines the effect or effects. A small intravenous dose causes generalized vasodilation (including the coronary and pulmonary circulation), whereas a larger dose is required to demonstrate negative chronotropic and dromotropic effects. Significant numbers of muscarinic receptors exist in these vascular beds despite the apparent absence of cholinergic nerve supply to most vessels. A second mechanism of vessel relaxation by acetylcholine is inhibition of norepinephrine release from adrenergic nerve terminals.
Acetylcholine decreases the rate of cardiac contraction, the velocity of conduction in the sinoatrial and atrioventricular nodes, and contractility (although not as marked as the increase produced by sympathetic stimulation) of the atria. In the sinoatrial node, acetylcholine causes membrane hyperpolarization, delaying resumption of the threshold potential and the ability to generate another action potential. This action slows the heart rate. Although the duration of the action potential and the effective refractory period are increased, the rate of conduction through atrial myocardium is unchanged. In the atrioventricular node, acetylcholine decreases conduction velocity and increases the effective refractory period. This decrease in nodal conduction usually accounts for the complete heart block seen when large amounts of cholinergic agonists are given. In the ventricle, acetylcholine decreases the Purkinje system automaticity, thereby increasing the fibrillation threshold. In the heart, presynaptic and postsynaptic muscarinic receptors are involved in these effects. Acetylcholine inhibits adrenergic stimulation of the heart presynaptically by inhibiting the release of norepinephrine from sympathetic nerve endings and postsynaptically by opposing the effects of catecholamines on the myocardium.
Parasympathetic activation has many effects outside the cardiovascular system. Acetylcholine stimulates the chemoreceptors of the carotid and aortic bodies. Cholinergic stimulation causes smooth muscle constriction, including that of bronchial walls. In the gastrointestinal and genitourinary tracts, smooth muscle in the walls constricts, but sphincter muscles relax. Topically administered acetylcholine constricts the smooth muscle of the iris, causing miosis.
Signs and symptoms of cholinergic overload reflect all these effects, with nausea and vomiting, intestinal cramps, belching, urination, and urgent defecation. All parasympathetic glands are stimulated to produce secretions, including the lacrimal, tracheobronchial, salivary, digestive, and exocrine glands.
In addition to the pharmacologic effects of acetylcholine that are mediated by the parasympathetic nervous system, acetylcholine has a significant effect on blood vessels, dilating virtually all vessels in vivo. In 1980, Furchgott and Zawadzki[36] observed that blood vessels with intact endothelium dilated when acetylcholine was applied. If the endothelial cells were damaged, the vessels constricted. Endothelial cells respond to acetylcholine stimulation by producing one or more endothelium-derived relaxing factors (EDRFs). [37] It now appears that the endothelial cells have receptors to numerous agonists, including serotonin, adenosine, histamine, and catecholamines ( Fig. 16-8 ). The radical NO is the first identified EDRF. It is produced by the endothelial cells in minute quantities, but it acts to relax vascular smooth muscle cells and to limit or modify the actions of many vasoconstrictors. Nitroglycerin is degraded to NO in the vascular myocyte. The mechanism of this vasodilator action is mediated through activation of guanylate cyclase by means of a protein phosphorylation cascade to phosphorylate actinomyosin directly. When the endothelium is damaged, as in atherosclerosis, production of EDRF diminishes and constriction increases. This change explains why patients with damaged or diseased vessels react differently.
The biology of NO appears to be important.[38] This simple compound, the mechanism of whose storage and release is still poorly understood, plays a prominent role in signal transduction and is a neurotransmitter in the gut and cerebellum (N-methyl-D-aspartate [NMDA] induces release of NO in the cerebellum).[39] [40] NO is produced during the conversion of arginine to citrulline by a class of enzymes known as NO synthases (NOS). Five isoforms of this family of enzymes have been identified, and the most common form, found in the cerebellum, has
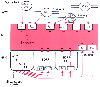 Figure 16-8
Schematic representation of potential modes of regulation
of vascular tone by endothelial cell-related mechanisms. Norepinephrine (NA), adenosine
triphosphate (ATP), calcitonin gene-related peptide (CGRP), substance P (SP), and
vasoactive intestinal polypeptide (VIP) can be released from nerves in the adventitia
(ADV) to act on their respective receptors in the media (MED) to cause vasoconstriction
or vasodilation. ATP, acetylcholine (ACh), 5-hydroxytryptamine (5-HT), and SP released
from endothelial cells (END) by shear stress or hypoxia act on their receptors on
endothelial cells to cause release of endothelium-derived relaxing factors (EDRF)
or prostaglandins (PG), which act on smooth muscle to cause relaxation. In areas
denuded of endothelial cells, opposite effects may be produced by receptors on the
smooth muscle. α, noradrenaline receptor; M, muscarinic receptor; P2x
,
P2x
-purinoceptor; P2y
, P2y
-purinoceptor. (From
Lincoln J, Burnstock G: Neural-endothelial interactions in control of local blood
flow. In Warren J [ed]: The Endothelium: An Introduction
to Current Research. New York, Wiley-Liss, 1990, p 21.)
Figure 16-8
Schematic representation of potential modes of regulation
of vascular tone by endothelial cell-related mechanisms. Norepinephrine (NA), adenosine
triphosphate (ATP), calcitonin gene-related peptide (CGRP), substance P (SP), and
vasoactive intestinal polypeptide (VIP) can be released from nerves in the adventitia
(ADV) to act on their respective receptors in the media (MED) to cause vasoconstriction
or vasodilation. ATP, acetylcholine (ACh), 5-hydroxytryptamine (5-HT), and SP released
from endothelial cells (END) by shear stress or hypoxia act on their receptors on
endothelial cells to cause release of endothelium-derived relaxing factors (EDRF)
or prostaglandins (PG), which act on smooth muscle to cause relaxation. In areas
denuded of endothelial cells, opposite effects may be produced by receptors on the
smooth muscle. α, noradrenaline receptor; M, muscarinic receptor; P2x
,
P2x
-purinoceptor; P2y
, P2y
-purinoceptor. (From
Lincoln J, Burnstock G: Neural-endothelial interactions in control of local blood
flow. In Warren J [ed]: The Endothelium: An Introduction
to Current Research. New York, Wiley-Liss, 1990, p 21.)
Endothelial cells control the circulation in addition to producing NO. Endothelial cells metabolize many vasoactive amines, convert angiotensin I to angiotensin II, and secrete NO, prostacyclin, and the vasoconstrictive peptide endothelin-1 (ET-1). NO, ET-1, and prostacyclin are local hormones released by endothelial cells to influence their immediate microenvironment. Prostacyclin and NO relax underlying smooth muscle on the abluminal side, whereas in the lumen, they act separately or in concert to prevent platelets from clumping onto the endothelium. There is clear synergism between the anti-aggregating effect of prostacyclin and subthreshold concentrations of NO. Substances that activate prostacyclin generally stimulate NO release. Shear stress increases NO as an adaptive mechanism to dilate the circulation actively. NO works by the guanylate cyclase mechanism; prostacyclin works by activation of adenylate cyclase. Although they work in concert, they activate different second messengers.[45]
Although prostacyclin and NO are short-lived vasodilators, injection of the vasoconstrictor ET-1 causes a powerful and long-lasting pressor effect. ET-1, which consists of 21 amino acids bound by two disulfide bonds,
NO, prostacyclin, and ET-1 appear to be operative in local control of the circulation. The prostacyclin system may be a mechanism reserved to reinforce the NO system in the presence of endothelial damage. Acting together, these two dilators are a strong defense mechanism against intravascular thrombosis. ET-1 can be produced locally in response to trauma such as wounds to the vessel wall. Long-term functions of these local hormones are of considerable interest in the pathophysiology of many disease states, including septic shock, pulmonary hypertension, and renal failure.[47] [48] [49]
|
|
|
|
|
|
|
|
|
|
|
|
|