|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In the 1970s, Ali and coauthors[560] [561] described a TOF ratio of 0.60 as being indicative of adequate recovery of neuromuscular strength. As described in the section "Monitoring Neuromuscular Function," with a TOF ratio of 0.6 to 0.7, patients will have, on average, a vital capacity of 55 mL/kg, a negative inspiratory force of 70 cm H2 O, and a peak expiratory flow of 95% of control values. This degree of recovery should allow for normal respiratory function[57] and maintenance of a patent airway. More recently, though, a TOF ratio of 0.70 in unanesthetized volunteers has been associated with difficulty speaking and swallowing, weakness of the facial musculature, visual disturbances, and an inability to sit up without assistance.[54] Administration of small doses of nondepolarizing neuromuscular blockers (one tenth of an intubating dose) as a defasciculating or precurarizing dose may cause appreciable decreases in the TOF response.[280] [562] These small doses of neuromuscular blockers may be associated with general discomfort, malaise, difficulty swallowing, ptosis, and blurred vision. Recent studies in volunteers have shown that TOF ratios of 0.6 to 0.7 are associated with decreased upper esophageal tone and a decrease in coordination of the esophageal musculature during swallowing.[563] [564] Fluoroscopic study of these individuals demonstrated significant pharyngeal dysfunction resulting in a fourfold to fivefold increase in the risk of aspiration. With recovery of the TOF ratio to 0.9, esophageal tone and pharyngeal coordination return toward baseline.
Residual paralysis also decreases the hypoxic ventilatory drive ( Fig. 13-27 ).[565] [566] This effect appears to be due to inhibition of the carotid body neural response to hypoxia.[567] Vecuronium decreases carotid sinus nerve activity in response to hypoxia in a dose-related fashion, presumably through its interaction with neural nicotinic receptors.
After the administration of nondepolarizing neuromuscular blocking drugs, it is essential to ensure adequate return of normal neuromuscular function. Whether that degree of recovery is a TOF ratio of 0.7, 0.8, or 0.9 is an area of debate.[568] Certainly, the clinician's ability to quantify the degree of residual neuromuscular block is limited (as described in the section "Monitoring Neuromuscular Function"). Kopman and colleagues' work in volunteers demonstrated that when they could oppose their incisors to retain a tongue depressor, their TOF ratio was, on average, 0.8 and at least 0.68.[54] This test of muscle strength, however, would be of limited usefulness in an intubated patient.
Recovery from muscle relaxation caused by nondepolarizing neuromuscular blockers is dependent on several factors. Primarily, it depends on an increase in the acetylcholine concentration relative to that of the relaxant to overcome the competitive neuromuscular block. The relative increase in acetylcholine concentration depends first on the ongoing movement of relaxant from the motor end plate into the central circulation and then on its elimination from the circulating blood volume so that it is not free to move into the synaptic cleft. Ultimately, recovery depends on elimination of the neuromuscular blocker from the body. Neuromuscular blockers may be eliminated from the body through a host of mechanisms, including excretion as unchanged drug in urine, metabolism in the liver, enzymatic hydrolysis, and chemical breakdown. Although it has never been specifically examined, several manuscripts have, through their ranges
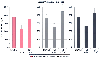 Figure 13-27
Hypoxic ventilatory response (HVR) before (control),
during steady-state infusion (train-of-four [TOF] ratio = 0.07) of atracurium, pancuronium
and vecuronium, and after recovery (TOF > 0.90). Data are presented as means
± SD. *P < .01. (Redrawn
from Eriksson LI: Reduced hypoxic chemosensitivity in partially paralysed man.
A new property of muscle relaxants? Acta Anaesthesiol Scand 40:520–523, 1996.)
Figure 13-27
Hypoxic ventilatory response (HVR) before (control),
during steady-state infusion (train-of-four [TOF] ratio = 0.07) of atracurium, pancuronium
and vecuronium, and after recovery (TOF > 0.90). Data are presented as means
± SD. *P < .01. (Redrawn
from Eriksson LI: Reduced hypoxic chemosensitivity in partially paralysed man.
A new property of muscle relaxants? Acta Anaesthesiol Scand 40:520–523, 1996.)
Several factors in addition to coexisting disease will have an impact on the speed of spontaneous recovery of neuromuscular function. The presence of volatile anesthetics will potentiate any existing neuromuscular block and, presumably, render recovery more prolonged.[572] If the anesthesiologist observes no or minimal recovery of neuromuscular function in the presence of a volatile anesthetic, discontinuing or decreasing the concentration of inhaled anesthetic being administered should augment recovery of neuromuscular function.
As will be discussed later, acidosis, hypokalemia, hypothermia, and concomitant medications will all potentiate residual neuromuscular blockade and render pharmacologic antagonism more difficult.
Anticholinesterases act by inhibiting the enzyme acetylcholinesterase. Acetylcholinesterase (enzyme classification 3.1.1.7) is a type B carboxylesterase. At the neuromuscular junction, it occurs in the asymmetric or A12 form, which consists of three tetramers of catalytic subunits covalently linked to a collagen-like tail. [573] Acetylcholinesterase has a powerful catalytic capacity.[29] It can catalyze 4000 molecules of acetylcholine per active site per second.[29] Nearly half of the released acetylcholine is hydrolyzed across the synaptic cleft before reaching nAChRs. The active site lies deep inside the enzyme protein.[574] For a detailed account of this enzyme, see Soreq and Seidman.[575]
The active surface of acetylcholinesterase is best viewed as having two sites: (1) the anionic site, which is concerned with binding and orienting the substrate molecule, and (2) the esteratic site, which is responsible for the hydrolytic process.[576] A second "anionic" site, known as the "peripheral" anionic site, was also proposed.[577]
Three anticholinesterases, neostigmine, edrophonium, and pyridostigmine, are used to antagonize residual neuromuscular blockade. They exert their effect primarily by increasing the concentration of acetylcholine at the motor end plate by inhibiting acetylcholinesterase. Neostigmine and pyridostigmine are oxydiaphoretic (acid transferring) inhibitors of acetylcholinesterase. Neostigmine and pyridostigmine transfer a carbamate group to the acetylcholinesterase, which forms a covalent bond at the esteratic site. Edrophonium binds to the anionic site on acetylcholinesterase by electrostatic attraction and to the esteratic subsite by hydrogen bonding.[578] In addition, anticholinesterases may also increase the release of acetylcholine from presynaptic nerve terminals, block neural potassium channels, and have a direct agonist effect.[27] Details of the mechanisms of action of these anticholinesterases have been described in review articles.[579] [580]
|
|
|
|
|
|
|
|
|
|
|
|
|