|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Much research has been conducted to delineate the mechanisms of action of barbiturates on the CNS, but with the exception of their action on the GABAA receptor, these mechanisms remain largely unknown.[307] [308] The selectivity of action of barbiturates on CNS neurophysiologic systems has been grouped into two general categories: (1) enhancement of the synaptic actions of inhibitory neurotransmitters and (2) blockade of the synaptic actions of excitatory neurotransmitters.[309] GABA is the principal inhibitory neurotransmitter in the mammalian CNS, and the GABAA receptor is the only site that has been proved to be involved in barbiturate-induced anesthesia.[308] The GABAA receptor is a chloride ion channel that is composed of at least five subunits with specific sites of action for GABA, barbiturates, benzodiazepines, and other molecules.[285] Binding of barbiturate to the GABAA receptor both enhances and mimics the action of GABA by increasing chloride conductance through the ion channel, causing hyperpolarization of the cell membrane, and thus increasing the threshold of excitability of the postsynaptic neuron. [310] At low concentrations, barbiturates enhance the effects of GABA by decreasing the rate of dissociation of GABA from its receptor and thus increasing the duration of GABA-activated chloride ion channel opening. This enhancement of the action of GABA is thought to be responsible for the sedative-hypnotic effects of barbiturates. At higher concentrations, barbiturates directly activate chloride channels without binding of GABA, thereby acting as the agonist themselves. The GABA-mimetic effect at slightly higher concentrations may be responsible for what is termed "barbiturate anesthesia."[308] [311]
Barbiturates also inhibit the synaptic transmission of excitatory neurotransmitters such as glutamate and acetylcholine.[308] The actions of barbiturates in blocking excitatory CNS transmission are specific for synaptic ion channels. Although these alternate sites have received much attention, their exact role, if any, in the anesthetic effects of barbiturates remains uncertain. [307] [312]
Barbiturates, like other CNS depressants, have potent effects on cerebral metabolism. Several studies in the 1970s demonstrated the effect of barbiturates to be a dose-related depression in CMRO2 , which produces progressive slowing of the EEG, a reduction in the rate of ATP consumption, and protection from incomplete cerebral ischemia.[313] [314] [315] The relationship of depressed metabolism to drug dosage was shown in dogs in which circulation at high thiopental doses was preserved by an extracorporeal circulation pump.[316] When the results of the EEG became isoelectric, a point at which cerebral metabolic activity is roughly 50% of baseline,[317] no further decrements in CMRO2 occurred. These findings support the hypothesis that metabolism and function are coupled. However, it must be noted that it is the portion of metabolic activity concerned with neuronal signaling and impulse traffic that is reduced by barbiturates, not the portion corresponding to basal metabolic function. The only way to suppress the baseline metabolic activity associated with cellular activity is through hypothermia.[317] Thus, the effect of barbiturates on cerebral metabolism is maximized at a 50% depression of cerebral function in which less oxygen is required as CMRO2 is diminished, with all metabolic energy left for the maintenance of cellular integrity.
With the reduction in CMRO2 comes a parallel reduction in cerebral perfusion, which is seen as decreased cerebral blood flow (CBF) and ICP. With reduced CMRO2 , cerebral vascular resistance increases and CBF decreases. [318] The ratio of CBF to CMRO2 is unchanged. Thus, the reduction in CBF after the administration of barbiturates causes a concurrent decrease in ICP. Furthermore, even though mean arterial pressure decreases, barbiturates do not compromise overall cerebral perfusion pressure because cerebral perfusion pressure equals mean blood pressure minus ICP. In this relationship, ICP decreases to a greater extent relative to the decrease in mean arterial pressure after barbiturate use, thus preserving cerebral perfusion pressure.
Barbiturates produce the clinical effects of sedation and sleep. Sufficient doses produce a CNS depression that is
Barbiturates produce CNS effects when they cross the blood-brain barrier. Several well-known factors help determine the rapidity with which a drug enters the cerebrospinal fluid (CSF) and brain tissue,[299] including the degree of lipid solubility, degree of ionization, level of protein binding, and the plasma drug concentration.
Drugs with high lipid solubility and a low degree of ionization cross the blood-brain barrier rapidly and produce a fast onset of action.[308] Most barbiturates exist in a nonionized form. The degree of lipid solubility of the nonionized form of the drug works in conjunction with the amount of drug that exists in the nonionized form (reflection of pKa ) to determine the rapidity with which a drug crosses the blood-brain barrier. Thiopental and methohexital are more lipid soluble than pentobarbital, which corresponds clinically to the more rapid onset of action of thiopental and methohexital than pentobarbital. [320] [321]
Only the nonionized form of a drug can directly traverse the cellular membranes. Thiopental has a pKa of 7.6. Therefore, approximately 50% of thiopental is nonionized at physiologic pH, which accounts in part for the rapid accumulation of thiopental in the CSF after intravenous administration.[322] Methohexital is 75% nonionized at pH 7.4, a fact that may explain the slightly more rapid effect of this drug than that of thiopental. As pH decreases, for example, with poor perfusion, barbiturates have a larger proportion of nonionized drug available to cross the blood-brain barrier. [321] [322] Obvious clinical consequences result: patients with acidosis require the administration of less barbiturate, and patients with alkalosis might require an increased dose to achieve the same level of anesthesia or sedation.
Protein binding also affects the onset of action in the CNS. Barbiturates are highly bound to albumin and other plasma proteins. Because only unbound drug (free drug) can cross the blood-brain barrier, an inverse relationship exists between the degree of plasma protein binding and the rapidity of drug passage across the blood-brain barrier.[302] Drugs have different degrees of protein binding, and in general, the thiobarbiturates are more highly bound than the oxybarbiturates.[323] The degree of protein binding of a drug is influenced by the physiologic pH and disease states that alter the absolute amount of protein. Most barbiturates tend to experience peak protein binding at or around pH 7.5, and slightly fewer drugs have most bound at a more acidic or basic pH. Disease states such as hepatic cirrhosis or chronic renal disease may reduce the total availability of plasma albumin, which could decrease the absolute amount of protein available for binding.[285] However, this situation may be of little clinical significance because most patients have many more protein molecules than drug molecules. Another possible consideration is whether other drugs that are highly protein bound can displace barbiturates from albumin. Again, such displacement is possible in theory, although an important clinical effect is unlikely to occur.
The final factor governing the rapidity of drug penetration of the blood-brain barrier is the plasma drug concentration. Simply because of the concentration gradient, higher drug concentrations in plasma produce greater amounts of drug that diffuses into the CSF and brain. The two primary determinants of the plasma concentration are the dose administered and the rate (speed) of administration. For example, as the dose of thiopental over the same time is increased, an increased percentage of patients are anesthetized.[324] As regards the absolute dose, 2 mg/kg produced anesthesia in 20% of patients, whereas a dose of 2.4 mg/kg produced anesthesia in 80% of patients. Similarly, the speed of injection influences the effect of thiopental.[325] When a fixed initial dose of thiopental of approximately 2.75 mg/kg was administered, a significantly (P <.001) smaller amount of drug was required to produce anesthesia when the dose was delivered over a period of 5 seconds as opposed to 15 seconds. Thus, the dose and rate of intravenous administration can profoundly affect the onset of barbiturate CNS effect.
Because brain and plasma concentrations are in equilibrium, factors that determine the rate of onset of barbiturate effect also affect their termination. Thus, lipid solubility, the degree of ionization, and the CSF drug concentration affect the movement of drugs from the CSF to plasma. Protein binding is less important because protein is not usually found in CSF, although there is certainly protein in the brain to which barbiturates may be bound. As plasma levels decrease, drug levels in the brain and CSF decrease. The most important factors in the termination of drug effect are those that govern plasma disappearance of the drug. These factors are generally divided into a rapid redistribution phase and a slow metabolic and second redistribution phase. In a classic pharmacologic study, Brodie and coworkers conclusively demonstrated that awakening from thiopental occurred because the plasma level rapidly declined.[326] They further demonstrated that the cause of the rapid plasma decay of thiopental was not metabolism of the drug but rather redistribution of the drug to other tissues throughout the body (lean tissue). The role of slow-phase distribution to adipose tissue and elimination and clearance from plasma by metabolism was elucidated later.[301] [323] The relationship of the plasma drug level to the onset and termination of effect as it relates to drug redistribution is illustrated in Figure 10-9 . As time passes, the blood level of barbiturate decreases and the drug is taken up by less well perfused tissues (first muscle and then fat). In addition, the rate of metabolic clearance is constant; the liver biotransforms a constant proportion or fraction (first-order kinetics) of the drug from blood. Clinically, patients awake from a single dose of thiopental 5 to 10 minutes after administration because the drug
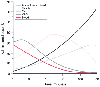 Figure 10-9
After delivery of an intravenous bolus, the percentage
of thiopental remaining in blood rapidly decreases as drug moves from blood to body
tissues. The time to attainment of peak tissue levels is a direct function of tissue
capacity for barbiturate relative to blood flow. Thus, a larger capacity or smaller
blood flow is related to a longer time to a peak tissue level. Initially, most thiopental
is taken up by the vessel-rich group (VRG) of tissues because of their high blood
flow. Subsequently, drug is redistributed to muscle and to a lesser extent to fat.
Throughout this period, small but substantial amounts of thiopental are removed
and metabolized by the liver. Unlike removal by tissues, this removal is cumulative.
Note that the rate of metabolism equals the early rate of removal by fat. The sum
of this early removal by fat and metabolism is the same as the removal by muscle.
(Redrawn from Saidman LJ: Uptake, distribution and elimination of barbiturates.
In Eger EI [ed]: Anesthetic Uptake and Action.
Baltimore, Williams & Wilkins, 1974.)
Figure 10-9
After delivery of an intravenous bolus, the percentage
of thiopental remaining in blood rapidly decreases as drug moves from blood to body
tissues. The time to attainment of peak tissue levels is a direct function of tissue
capacity for barbiturate relative to blood flow. Thus, a larger capacity or smaller
blood flow is related to a longer time to a peak tissue level. Initially, most thiopental
is taken up by the vessel-rich group (VRG) of tissues because of their high blood
flow. Subsequently, drug is redistributed to muscle and to a lesser extent to fat.
Throughout this period, small but substantial amounts of thiopental are removed
and metabolized by the liver. Unlike removal by tissues, this removal is cumulative.
Note that the rate of metabolism equals the early rate of removal by fat. The sum
of this early removal by fat and metabolism is the same as the removal by muscle.
(Redrawn from Saidman LJ: Uptake, distribution and elimination of barbiturates.
In Eger EI [ed]: Anesthetic Uptake and Action.
Baltimore, Williams & Wilkins, 1974.)
Findings of pharmacokinetic analysis illustrate that awakening from thiopental is related to rapid redistribution.[42] As previously mentioned, awakening is independent of the dose administered, except in the case of continuous infusion, which prolongs recovery. Awakening may be delayed in older patients because of either increased CNS sensitivity, alterations in metabolism, or a decreased central volume of distribution relative to younger adults.[327] The initial volume of distribution is less in elderly patients than young patients, which explains their lower dose requirement for the onset of EEG and hypnotic effects. [327] Pediatric patients (younger than 13 years) seem to have a greater rate of total clearance and a shorter rate of plasma thiopental clearance than adults do, which theoretically might result in earlier awakening, especially after multiple doses of the drug.[328]
Thiopental and methohexital are not very different with regard to distribution, which may explain the similar wake-up time with these drugs. There is, however, a difference in the rate of total-body clearance, with methohexital being higher. This disparity could explain the difference found in the psychomotor skills of patients and the earlier full recovery after methohexital. Sensitive tests of psychomotor skills tend to show better early performance after methohexital than after thiopental use. A driving test, however, reveals abnormal skills for as long as 8 hours after anesthesia, thus suggesting that despite plasma clearance, residual CNS impairment lasts for about 1 day.[329] Regardless of these residual effects, methohexital is cleared more rapidly than thiopental, which explains why methohexital is preferred for use by some clinicians when rapid awakening is desirable, such as in outpatient anesthesia. This prolongation of both early and late recovery by barbiturates is why they have largely been replaced by propofol.
|
|
|
|
|
|
|
|
|
|
|
|
|