PATHOPHYSIOLOGY AND MOLECULAR BIOLOGY
MH is a myopathy, usually subclinical, that features an acute
loss of control of intracellular calcium ions (Ca2+
). Normal muscle contraction
is initiated at the neuromuscular junction (i.e., the motor end plate). Acetylcholine
is released from the terminals of motor neurons and diffuses a short distance to
the postsynaptic membrane, where binding to nicotinic cholinergic receptors triggers
a wave of depolarization referred to as an excitatory postsynaptic
potential (EPSP) that leads to action potentials that propagate to transverse
tubules (T tubules). The T tubules act as conduits to bring action potentials deep
within the myofibrils, where their excitatory signal is
transduced to the junctional face of sarcoplasmic reticulum (SR) within the muscle
cells to initiate release of Ca2+
stored within the SR terminal cisternae.
In skeletal muscle, the release of SR Ca2+
is an essential step for contraction.
The whole process, from T-tubule depolarization to release of SR Ca2+
,
is called excitation-contraction (EC) coupling. Knowledge of the molecular events
contributing to EC coupling is essential to understanding the cause of MH.
Skeletal EC coupling begins deep within the T-tubule membrane
at high-density, voltage-gated L-type Ca2+
channels, historically labeled
dihydropyridine receptors (DHPRs). The DHPR possesses
an integral membrane voltage sensor whose function is to respond to T-tubule depolarization
and initiate long-range conformational changes within the Ca2+
channel
complex. Voltage-dependent activation of DHPR opens an integral Ca2+
-selective
conductance path that permits entry of small amounts of Ca2+
into the
muscle cell. The entry of Ca2+
into the skeletal myotube is not necessary
for engaging skeletal-type EC coupling. Rather, one of the DHPR subunits (α1S
-subunit)
provides physical links between DHPRs within T tubules and Ca2+
release
channels within junctional SR. Skeletal muscle expresses a specific type of Ca2+
release channel called the skeletal isoform, or RYR1.
Skeletal EC coupling is the result of physical coupling between α1S
-subunits
and RYR1 at specialized "triadic" regions where the T-tubule membrane comes in close
apposition to junctional SR and does not depend on the influx of Ca2+
.
After EC coupling is initiated, the free, ionized, unbound intracellular Ca2+
concentration within
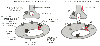 Figure 29-1
The key ion channels involved in neuromuscular transmission
and excitation contraction coupling. Nerve impulses arriving at the nerve terminal
activate voltage-gated Ca2+
channels (1). The resulting increase in cytoplasmic
Ca2+
concentration is essential in exocytosis of acetylcholine. Binding
of acetylcholine to postsynaptic nicotinic cholinergic receptors activates an integral
nonselective cation channel, which depolarizes the sarcolemmal membrane (2). Depolarizing
the sarcolemma to threshold activates voltage-gated Na+
channels (3),
which propagate action potential impulses deep into the muscle through the transverse
tubule system. Within the transverse tubule system, L-type voltage-gated Ca2+
channels sense membrane depolarization and undergo a conformational change (4).
A physical link between the α1
-subunit and the ryanodine receptor
is thought to transfer the signal to sarcoplasmic reticulum to induce the release
of stored Ca2+
(5). (Adapted from Alberts B, Bray D, Lewis J,
et al: Molecular Biology of the Cell, 3rd ed. New York, Garland Press, 1994.)
Figure 29-1
The key ion channels involved in neuromuscular transmission
and excitation contraction coupling. Nerve impulses arriving at the nerve terminal
activate voltage-gated Ca2+
channels (1). The resulting increase in cytoplasmic
Ca2+
concentration is essential in exocytosis of acetylcholine. Binding
of acetylcholine to postsynaptic nicotinic cholinergic receptors activates an integral
nonselective cation channel, which depolarizes the sarcolemmal membrane (2). Depolarizing
the sarcolemma to threshold activates voltage-gated Na+
channels (3),
which propagate action potential impulses deep into the muscle through the transverse
tubule system. Within the transverse tubule system, L-type voltage-gated Ca2+
channels sense membrane depolarization and undergo a conformational change (4).
A physical link between the α1
-subunit and the ryanodine receptor
is thought to transfer the signal to sarcoplasmic reticulum to induce the release
of stored Ca2+
(5). (Adapted from Alberts B, Bray D, Lewis J,
et al: Molecular Biology of the Cell, 3rd ed. New York, Garland Press, 1994.)
the relaxed muscle cell increases from 10-7
M to about 5 ×
10−5
M. The increase in Ca2+
removes the
troponin inhibition from the contractile proteins, resulting in muscle contraction.
Intracellular Ca2+
pumps (i.e., sarcoplasmic/endoplasmic reticulum Ca2+
-ATPase
[SERCA] pumps) rapidly reaccumulate Ca2+
back into the SR, and relaxation
occurs when the concentration is restored to less than mechanical threshold. Contraction
and relaxation require adenosine triphosphate (ATP); both are energy-related processes
that consume ATP ( Fig. 29-1
).
Clinical and laboratory data for swine and humans indicate decreased
control of intracellular Ca2+
, resulting in a release of free, unbound,
ionized Ca2+
from storage sites that normally maintain muscle relaxation.
Aerobic and anaerobic forms of metabolism increase to provide added ATP to drive
the Ca2+
pumps that maintain Ca2+
homeostasis in SR and mitochondria
and across the sarcolemma in extracellular fluid. Virtually all of these reactions
are exothermic (i.e., they produce heat). Rigidity occurs when unbound myofibrillar
Ca2+
approaches the contractile threshold. Dantrolene is therapeutic
because it reduces Ca2+
release from the SR without altering Ca2+
reuptake.
Molecular Events in Excitation-Contraction Coupling
Understanding the mechanisms underlying MH requires a more detailed
description of the process of EC coupling, the process by which skeletal muscle transforms
a chemical signal in the form of a neurotransmitter at the surface
of the fiber into muscle contraction.[17]
[18]
[19]
Figure
29-1
depicts a neuromuscular junction in which an efferent motor neuron
synapses with muscle fibers to form a motor end plate. Membrane depolarization at
the nerve terminal activates voltage-dependent Ca2+
channels on the presynaptic
membrane. Most members of the family of voltage-dependent Ca2+
channels
are composed of five subunits: α1
, α2
, β,
γ, and δ. Although subunits α2
, β, γ, and
δ contribute important membrane targeting and modulatory functions, it is the
larger α1
-subunit that performs essential functions of voltage sensing
and conduction of Ca2+
. Specifically, it is the N-type channel (i.e.,
CaV
2.1 in International Union of Pharmacology nomenclature, α1A
in alternate nomenclature) that is primarily responsible for depolarized induced
Ca2+
entry into motor nerve terminals. Rise of cytosolic Ca2+
concentrations within the nerve terminals initiates a process of vesicle migration
and fusion that leads to exocytosis of acetylcholine stored within the synaptic vesicles.
Simultaneous release of thousands of quanta of acetylcholine results in EPSPs.
Acetylcholine binds to nicotinic acetylcholine receptors, which are nonselective
cation channels, and activates inward current (primarily carried by sodium ions),
thereby depolarizing the muscle cells. When EPSPs sum to threshold, action potentials
are propagated from the sarcolemma to the T tubule. Acetylcholinesterase in the
synaptic cleft catalyzes rapid breakdown of acetylcholine; rapid removal from the
cleft enables the motor unit to be ready for another stimulus within a few milliseconds.
Within the T-tubule membrane of skeletal muscle, a highly homologous
relative of the N-type channels, the L-type voltage-gated Ca2+
channel
(CaV
1.1 or α1S
), or DHPR, is enriched within the T-tubule
membrane. Three chemical
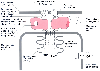 Figure 29-2
Schematic representation of the triad junction of skeletal
muscle shows the junctional foot protein (ryanodine receptor [RyR1]) and its associated
proteins. In skeletal muscle, the α1S
-subunit of the dihydropyridine
receptor (DHPR) participates in excitation-contraction coupling. These physical
links transmit essential signals across the narrow gap of the triadic junction that
activate RyR1 and release Ca2+
from the sarcoplasmic reticulum. (Adapted
from Pessah IN, Lynch C III, Gronert GA: Complex pharmacology of malignant hyperthermia.
Anesthesiology 84:1275, 1996.)
Figure 29-2
Schematic representation of the triad junction of skeletal
muscle shows the junctional foot protein (ryanodine receptor [RyR1]) and its associated
proteins. In skeletal muscle, the α1S
-subunit of the dihydropyridine
receptor (DHPR) participates in excitation-contraction coupling. These physical
links transmit essential signals across the narrow gap of the triadic junction that
activate RyR1 and release Ca2+
from the sarcoplasmic reticulum. (Adapted
from Pessah IN, Lynch C III, Gronert GA: Complex pharmacology of malignant hyperthermia.
Anesthesiology 84:1275, 1996.)
classes of drugs used for controlling cardiovascular function—dihydropyridines,
phenylalkylamines, and benzothiazepines—are also capable of blocking DHPRs
by direct interaction with α1
-subunits. In skeletal muscle, it
is α1S
-DHPR that participates in EC coupling. Unique to skeletal
muscle is the highly ordered arrangement of α1S
-DHPRs into linear
arrays of clustered tetrads. Electron microscopic and immunocytochemical analyses
indicate that each α1S
-DHPR tetrad is close to (superimposed above)
a single RYR1 within the junctional face of SR terminal cisternae ( Fig.
29-2
). Because each functional RYR1 channel is composed of a tetramer
of four identical subunits, each α1S
-DHPR overlies a single RYR1
subunit. The relative restrictions imposed by the dimensions of α1S
-DHPR
tetrads and RYR1 tetramers permit only alternate RYR1 channels to pair with α1S
-DHPR
tetrads. In common with other voltage-gated ion channels, each α1S
-DHPR
possesses a stretch of amphipathic amino acids within the fourth α-helix (S4)
of each of four transmembrane domains that functions as a voltage sensor within the
T-tubule membrane. Membrane depolarization induces a discrete movement of charge
within the S4 segment of the α1S
-DHPR. A mechanical signal, thought
to be in the form of a conformational transition, is transmitted to the cytoplasmic
loop between repeats II and III of the α1S
-DHPR. Significant evidence
exists for a direct physical coupling between the II-III loop of α1S
-DHPR
and multiple noncontiguous regions within the large, hydrophilic cytoplasmic domain
of RYR1. Such physical links transmit essential signals across the narrow gap of
the triadic junction that activate RYR1 and release Ca2+
from SR. This
conformational coupling model is consistent with the nature of skeletal muscle EC
coupling,
which is independent of extracellular Ca2+
. However, after the initial
signal activates Ca2+
release from SR, Ca2+
-induced Ca2+
release appears to play a role in regulating the temporal and quantitative characteristics
of RYR1 activation. RYR1 also sends a retrograde signal to α1S
-DHPR,
enhancing its Ca2+
entry function. However, unlike Ca2+
entry
mediated by α1S
-DHRP, which is essential for cardiac EC coupling,
Ca2+
entry through α1S
-DHPR is neither essential nor
needed for engaging skeletal type EC coupling. Bidirectional signaling between α1S
-DHPR
and RYR1 in skeletal muscle appears to represent a fundamental mechanism involving
conformational coupling between sarcoplasmic/endoplasmic reticulum Ca2+
release channels and voltage- or store-operated Ca2+
entry (SOC) channels
within the surface membrane in a variety of mammalian cells.
After release into the sarcoplasm, Ca2+
is rapidly
removed through active transport by SERCA pumps located on junctional and longitudinal
SR. Calsequestrin inside the lumen of SR binds to Ca2+
and further enhances
Ca2+
loading within SR. Cytosolic Ca2+
is typically brought
back to basal nanomolar concentration within 30 msec of muscle contraction. The
rapid removal of cytosolic Ca2+
is essential for normal muscle relaxation
and requires rapid termination of Ca2+
efflux from SR. Aberrant termination
of RYR1 activity has emerged as a key underlying mechanism in MH susceptibility.