|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Persistent or repeated intermittent hepatic injuries can lead to hepatic cirrhosis. Alcoholism and chronic viral hepatitis (B and C) are the most common causes of cirrhosis in the United States.[340] Because of the large physiologic reserves of the liver, low-grade hepatic inflammation diseases (like hepatitis C) may remain subclinical while they are destroying much of the liver. Pathologic changes in the liver may include cellular hypertrophy, fatty metamorphosis, mitochondrial and lysosomal dysfunction, and disruption of transport and storage processes. Eventually, significant fibroplasia occurs, and, as hepatocytes become encircled by fibrous tissue, the liver becomes cirrhotic.[341] The progression of cirrhosis induces portal hypertension. Patients typically have a hyperdynamic circulation with extensive arteriovenous communications, and a decreased effective plasma volume. The characteristic clinical features of cirrhosis and portal hypertension include weakness, anorexia, nausea, vomiting, abdominal discomfort, jaundice, spider nevi, ascites, splenomegaly, a firm liver, esophageal varices, hepatorenal syndrome, and portosystemic encephalopathy. Advanced liver disease affects nearly every organ in the body and poses complex challenges to the health care team.[342] The multiple severe pathophysiologic derangements set the stage for devastating complications, including life-threatening hemorrhage, infection, renal failure, coma, and death.
Advanced liver disease often causes pulmonary dysfunction and hypoxemia.[343] [344] [345] Various pathologic changes, such as increased intrapulmonary arteriovenous
Portal hypertension combined with cirrhosis causes two major changes in renal function: avid sodium retention and decreased glomerular filtration rate (GFR). Renal function decreases progressively despite the absence of overt renal injury (e.g., no histopathology) or tubular dysfunction. The urinalysis typically is normal, and the urine sodium concentration is low. Prerenal disorders are chiefly responsible for the most severe renal complications of advanced liver disease—acute renal failure (ARF) and the hepatorenal syndrome.[29] [347] [348] [349]
In patients with advanced liver disease, poor renal perfusion is the major cause of both prerenal ARF and acute tubular necrosis (ATN). Renal hypoperfusion is usually the result of intravascular volume depletion, hypotension, or type 1 hepatorenal syndrome (HRS). Common causes of intravascular volume depletion include hemorrhage (e.g., variceal bleeding), ascites formation, sequestration of blood volume in the splanchnic vasculature, and increased loss of fluid from the kidneys (e.g., vigorous diuretic therapy) and gastrointestinal tract. Restoring renal blood flow—which involves identifying and treating the specific cause of the hypoperfusion—is the key to correcting prerenal ARF.
Proper fluid replacement is effective in most cases of "non-HRS" prerenal failure. ATN is usually a sequela of prolonged renal hypoperfusion (prerenal ARF), which eventually causes renal tubular ischemia. Other potential causes of ATN are nonsteroidal anti-inflammatory drugs and intravascular radiocontrast agents. There is no specific treatment for ATN; care is mainly supportive.[350] For type 1 HRS, when the expected short-term survival rate is low, liver transplantation is the preferred treatment. Type 2 HRS (the more stable form of HRS) usually occurs in patients with preserved hepatic function. Treatments that may transiently improve renal performance include (1) intravenous vasoconstrictors such as terlipressin, noradrenaline, and midodrine, in combination with octreotide; (2) molecular adsorbent recirculating system (MARS); and (3) transjugular intrahepatic portosystemic shunt (TIPS).[350]
Hematologic and hemostatic abnormalities are common in severe liver disease.[115] Anemia may occur because of plasma volume expansion, gastrointestinal bleeding, malnutrition, vitamin deficiencies, hemolysis, hypersplenism, or bone marrow depression. As the synthesis of vitamin K-dependent factors decreases, coagulopathies develop. The factor VII level must decrease by roughly 60% to 70% before the prothrombin time becomes prolonged. Thrombocytopenia and thrombopathy usually coexist with overt portal hypertension (ascites, splenomegaly). Causes of these abnormalities include splenic sequestration syndrome, bone marrow suppression (from alcohol or interferon or other medications), and immune-mediated platelet destruction (platelet-associated IgG). Dysfibrinogenemia (activation of fibrinolysis) is not uncommon. In this disorder, increases in fibrin degradation products and D-dimer occur but fibrinogen levels may be normal. The presence of overt disseminated intravascular coagulation, however, should prompt a rigorous pursuit of various reversible causes, such as endotoxemia.
Patients with cirrhosis and portal hypertension are at high risk for developing esophagogastric varices and portal hypertensive gastropathy. The prevalence of this latter condition parallels the severity of portal hypertension and liver dysfunction. Severe bleeding as a result of this gastropathy seldom occurs. [351] In contrast, hemorrhage from ruptured esophagogastric varices accounts for about one third of the deaths in this patient population. Generally, gastric varices bleed less often but more severely than esophageal varices. Variceal size is the most important predictor of variceal bleeding. The rates of variceal formation and enlargement parallel the degree of liver dysfunction. In patients with cirrhosis and portal hypertension, the incidence of variceal bleeding at 2 years ranges from 20% to 30%.[351] The first episode of variceal hemorrhage is acutely lethal in about 7% of cases; the 6-week mortality rate approaches 30%. Rebleeding is a major concern. Survivors of a variceal hemorrhage, if untreated, have a 60% risk of rebleeding within 2 years; the associated death rate is around 40% to 50%. Because of this poor prognosis, it is important to begin therapy promptly to prevent rebleeding.
Advanced liver disease produces several endocrine abnormalities. For example, increased plasma concentrations of growth hormone and glucagon contribute to insulin resistance. Decreased plasma concentrations of insulin-like growth factor 1 (IGF-1) adversely affect normal growth and development.[137] Gonadal dysfunction occurs in both men and women secondary to abnormal metabolism of sex hormones. Men undergo feminization, developing gynecomastia, testicular atrophy, infertility, and impotence. In women, oligomenorrhea, amenorrhea and infertility are common.
Hepatic encephalopathy is a complex neuropsychiatric syndrome, occurring in 50% to 70% of patients with cirrhosis.[29] The pathogenesis is complex and involves decreased hepatocellular function, reduced hepatic blood flow, and diversion of portal flow through extrahepatic collateral vessels (i.e., portacaval blood flow). Various chemical substances may contribute to the encephalopathy, including ammonia and various gut-derived molecules—short-chain fatty acids, mercaptans, phenols,
Advanced liver disease induces a hyperdynamic cardiovascular system characterized by increased cardiac output and low systemic vascular resistance.[353] Heart rate is often mildly increased, systemic arterial pressure is slightly decreased, and central filling pressures are normal. The increase in cardiac output leads to an increase in mixed venous O2 saturation and a decrease in the arteriovenous O2 content gap. Overall, the clinical and pathophysiologic changes resemble those of a peripheral arteriovenous fistula. These changes are mainly a result of widespread arteriovenous communications within the splanchnic organs, lungs, muscle, and skin. Vasodilatory substances, including glucagon, NO·, ferritin, and vasoactive intestinal polypeptide (VIP), may contribute to the high blood flow through these collateral vessels.[354] [355] [356] [357] [358] [359] [360]
Increases in endogenous vasodilators may also explain the decreases in cardiovascular responsiveness to physiologic (e.g., epinephrine) and pharmacologic vasoconstrictors in patients with advanced liver disease.[361] Laboratory investigations suggest that NO· could mediate the quintessential cardiovascular and renal changes of cirrhosis and portal hypertension.[362] [363] In animal models of chronic portal hypertension (partial portal vein ligation, CCl4 -induced cirrhosis), NO· induces cyclic guanosine monophosphate (cGMP) elevations that correlate directly with peripheral arterial dilation.[363] [364] Also, inhibition of NO· synthase (1) prevents the increase in cardiac output and the decrease in total peripheral resistance; (2) restores plasma aldosterone and arginine vasopressin levels; and (3) corrects the derangements of sodium and water excretion. Clinical investigations of healthy subjects and patients with cirrhosis have shown a positive correlation between the partial pressure of exhaled NO· and the cardiac index.[365]
The pathogenesis of portal hypertension includes one or more of the following influences: (1) increased pre-portal blood flow, (2) increased resistance to intrahepatic flow, and (3) increased flow through the portacaval collaterals. The classic "backward theory" proposes that proliferating fibrotic tissue induces cirrhosis and increases portal venous resistance, which causes portal pressure to rise. However, many observations are inconsistent with this theory. For example, experimental narrowing of the portal vein induces portal hypertension and the following effects: (1) increased mesenteric vascular resistance, (2) decreased mesenteric flow, (3) decreased splanchnic venous oxygen saturation, and (4) increased mesenteric arteriovenous oxygen content gap. This is the exact opposite of what happens in patients with cirrhosis and portal hypertension.
The "forward theory" addresses clinical and pathophysiologic features not well explained by the "backward theory."[366] According to the forward theory, endogenous vasodilators (e.g., NO·, glucagon, prostacyclin, adenosine) stimulate arteriovenous fistulae to form in the intestine and spleen. Splanchnic blood flow and cardiac output both increase, whereas portal venous flow decreases substantially. Hepatic arterial flow remains unchanged or may increase. The hepatic oxygen supply is preserved despite a decrease in total hepatic blood flow.
When volume receptors detect decreases in effective plasma volume, they activate the sympathetic nervous system.[367] This stimulates the kidney to release renin, which increases angiotensin II and aldosterone production. Increases in aldosterone and sympathoadrenal tone cause the renal tubules to avidly resorb sodium. There is an inverse relation between plasma norepinephrine level and blood flow to the kidney—suggesting a key role for sympathoadrenal activation in the renal retention of sodium. Vasoconstrictive effects of norepinephrine and angiotensin II redistribute intrarenal blood flow, which increases sodium uptake by the kidney. The kallikrein-kinin system also modulates sodium retention. Patients with cirrhosis and ascites have increased levels of endothelin, a potent vasoconstrictor and a likely contributor to the renal dysfunction. When cirrhosis and ascites coexist, vasodilatory prostaglandins are important for preserving renal blood flow. Inhibitors of prostaglandin synthesis decrease renal plasma flow and GFR and may therefore cause acute renal injury.
Patients with cirrhosis and portal hypertension develop edema, ascites, and increases in total body water because the kidneys avidly retain sodium ( Fig. 19-7 .[1] [368] [369] The pathogenesis of the sodium retention remains unclear. For instance, it is unknown whether severe liver disease mainly produces abnormal mediators that promote excessive sodium retention, or whether it decreases effective blood volume and therefore causes normal kidneys to avidly retain sodium.[367]
The first idea—the "overflow theory"—presumes that the kidney will hold on to sodium and water despite an overfilled intravascular compartment, whereas the second hypothesis—the "underfill theory"—simply reflects the normal homeostatic response to intravascular volume depletion (sodium and water retention). According to the overflow theory, sodium and water retention expands the intravascular volume and lowers plasma oncotic pressure. Expansion of the plasma volume increases portal hydrostatic pressure, which eventually causes portal hypertension. Portal hypertension and low oncotic pressure together cause edema and ascites. Proof for this theory will require identifying a primary pathogenic agent or process that causes the kidney to retain sodium when the plasma volume is increased.
According to the underfill theory, cirrhosis causes a decrease in the effective plasma volume, which stimulates
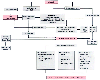 Figure 19-7
Schematic of pathways for cirrhosis-induced portal hypertension:
the forward and backward theories. Cirrhosis and portal hypertension induce circulatory
changes that decrease the effective blood volume. This activates volume receptors
and stimulates neurohumoral and intrarenal reflexes to decrease renal blood flow
and to increase renal retention of sodium. PVBF, portal venous blood flow; HABF,
hepatic arterial blood flow; THBF, total hepatic blood flow; A-V, arteriovenous;
PG's, prostaglandins; ADH, antidiuretic hormone; ANF, atrial natriuretic factor;
PAF, platelet activating factor. (Reprinted with permission from Mushlin
PS, Gelman S: Anesthesia and the liver. In Barash
PG, Cullen BF, Stoelting RK [eds]: Clinical Anesthesia, 4th ed. Philadelphia, Lippincott
Williams & Wilkins, 2001, p. 1088.)
Figure 19-7
Schematic of pathways for cirrhosis-induced portal hypertension:
the forward and backward theories. Cirrhosis and portal hypertension induce circulatory
changes that decrease the effective blood volume. This activates volume receptors
and stimulates neurohumoral and intrarenal reflexes to decrease renal blood flow
and to increase renal retention of sodium. PVBF, portal venous blood flow; HABF,
hepatic arterial blood flow; THBF, total hepatic blood flow; A-V, arteriovenous;
PG's, prostaglandins; ADH, antidiuretic hormone; ANF, atrial natriuretic factor;
PAF, platelet activating factor. (Reprinted with permission from Mushlin
PS, Gelman S: Anesthesia and the liver. In Barash
PG, Cullen BF, Stoelting RK [eds]: Clinical Anesthesia, 4th ed. Philadelphia, Lippincott
Williams & Wilkins, 2001, p. 1088.)
The mechanisms underlying the overflow and underfill theories are not mutually exclusive. Each could play a role in various stages of cirrhosis. For example, in the early stages, a primary defect in sodium excretion could play the more critical role, whereas with more advanced disease, a decrease in circulating plasma volume could be the more important cause.[368]
|
|
|
|
|
|
|
|
|
|
|
|
|