|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Many of our presumptions about cholinergic pharmacology are drawn from what is known about the neuromuscular junction, for which detailed electrophysiologic information on cholinergic transmission is readily available. Acetylcholine is synthesized intraneurally from acetyl coenzyme A and choline through the enzyme choline acetyl transferase in the synaptosomal mitochondria ( Fig. 16-17 ).[147] Despite the presence of this enzyme, choline is not made in the brain, but it appears to be transported. Sources of choline include dietary phospholipids, hepatic synthesis of phosphatidylcholine from dietary precursors such as ethanolamines, and choline released by hydrolysis of acetylcholine. Most choline originates in the liver. Choline is transported as
The neuromuscular junction includes the nerve ending, the muscle, and the synaptic cleft, which is a space between the nerve and the muscle. On the nerve or presynaptic side, the nerve ending contains many synaptic vesicles (i.e., quanta) that contain acetylcholine. On the muscle membrane, there are many infoldings of the postjunctional membrane. There are presynaptic release sites located opposite the junctional folds at the shoulders
 Figure 16-18
Acetylcholine (ACh) release, diffusion across the synaptic
cleft, binding to receptors on the end-plate membrane, and hydrolysis by acetylcholine
esterase (AChE) in the absence of blocking drugs. (From Stiller R: Neuromuscular
blocking agents. In Wingard L, Brody T, Larner J,
et al [eds]: Human Pharmacology: Molecular to Clinical. St. Louis, Mosby-Year
Book, 1991.)
Figure 16-18
Acetylcholine (ACh) release, diffusion across the synaptic
cleft, binding to receptors on the end-plate membrane, and hydrolysis by acetylcholine
esterase (AChE) in the absence of blocking drugs. (From Stiller R: Neuromuscular
blocking agents. In Wingard L, Brody T, Larner J,
et al [eds]: Human Pharmacology: Molecular to Clinical. St. Louis, Mosby-Year
Book, 1991.)
Acetylcholine is stored in vesicles approximately 300 µm in diameter, with each containing 10,000 molecules of the chemical. Central cholinergic transmission is different from the neuromuscular model in that few vesicles are present in the presynaptic terminal. These vesicles appear clear on electron microscopy.
The spontaneous release of the acetylcholine in one vesicle causes a miniature end-plate potential of 0.5 millivolts (mV), and 2000 channels open in the membrane of a muscle cell, permitting sodium influx and potassium efflux. Twelve thousand ions enter the postsynaptic cell each millisecond that each channel is open.
When a nerve impulse arrives at the presynaptic nerve terminal, it causes an influx of calcium ions across the membrane. This induces 100 to 300 synaptic vesicles to fuse with the presynaptic membrane at specific release sites at the active zone, resulting in the liberation of acetylcholine from vesicles into the synaptic cleft.
Although the original hypothesis proposed by Katz, of calcium as the exocytotic trigger, has stood the test of time, calcium entrance is necessary but not sufficient; the
Normally, in the absence of a nerve impulse, a series of miniature end-plate potentials (MEPPs) results from the spontaneous release of one quantum of acetylcholine (10,000 molecules, or the contents of one vesicle). Each opened channel produces a depolarization of 0.00022 mV. The MEPP involves 1500 ion channels being opened and causes a deflection of 0.5 mV. The EPP, which is summated, involves 500,000 ion channels opening and represents a depolarization of 50 to 100 mV.
Acetylcholine is an ester that hydrolyzes spontaneously in alkaline solutions into acetate and choline, neither of which has significant pharmacologic action. In vivo, the rate of hydrolysis is increased enormously by enzymatic catalysis. The two most important enzymes are acetylcholinesterase and butyrylcholinesterase.
Sometimes called tissue esterase or true esterase, acetylcholinesterase is a membrane-bound enzyme that is present in all cholinergic synapses, where it functions to destroy the neurotransmitter released from the nerve endings. Acetylcholinesterase is one of the most efficient of enzymes and destroys acetylcholine, terminating transmission within milliseconds after its release. The enzyme also is present in tissues that are not innervated, such as erythrocytes. Its function in these tissues is not known.
Butyrylcholinesterase, sometimes called plasma esterase or pseudocholinesterase, is a soluble enzyme that is made primarily in the liver and circulates in the blood. Its function in normal situations is not known; individuals who are genetically incapable of making the enzyme are normal in all other regards.
Of these two enzymes, acetylcholinesterase is the more important in terms of the function of acetylcholine. It is present in all cholinergic synapses, where it destroys neurally released acetylcholine, and it is the more efficient of the two enzymes. The substrate turnover is 2500 molecules per second, and one catalytic event lasts 40 µsec. Butyrylcholinesterase is important for the destruction of some cholinergic drugs, many of which are not destroyed by acetylcholinesterase.
Both forms of cholinesterase have been cloned. It is of more than passing interest that the first example of gene amplification in humans occurred in this enzyme system. The offspring of Israeli farmers exposed to insecticide expressed abnormal cholinesterase.[150] [151]
Traditionally, the catalytic part of the enzyme has been thought to contain two areas: an anionic site, which carries a strong negative charge, and an esteratic site, which contains electrophilic amino acids. It has been believed that acetylcholine during its hydrolysis is attracted to the enzyme because the negative charge of the anionic site attracts the positive charge in the quaternary nitrogen of acetylcholine. The ester group of acetylcholine aligns with the esteratic site of the enzyme. After an electrophilic attack on the molecule, the acetate link is transferred from the choline to an amino acid in the enzyme. The choline drifts away, leaving a covalently bound, acetylated enzyme. The acetate link is subsequently attacked and broken by a hydroxyl group from water. The acetate drifts away, and the regenerated enzyme is ready to interact with another molecule of acetylcholine.
However, the atomic structure of acetylcholinesterase is different from what had been previously thought, and our understanding of the binding of acetylcholine to its hydrolytic enzyme has altered.[152] The new model predicts that the quaternary ammonium binds to some of the 14 aromatic amino acids lining a deep gorge in the enzyme.
Inhibition of acetylcholinesterase prevents the destruction of acetylcholine in cholinergic synapses and can activate all cholinergic systems simultaneously. In addition to their therapeutic use, cholinesterase inhibitors are the active ingredients in insecticides and many nerve gases.
Traditionally, cholinergic receptors have been organized into two major subdivisions, nicotinic and muscarinic, that predict most clinical effects. Muscarinic receptors are present mostly in peripheral visceral organs; nicotinic receptors are found on parasympathetic and sympathetic ganglia and on the neuromuscular junctions of skeletal muscle.
Although these two structurally and functionally distinct classes of receptors have significantly different responses to acetylcholine, the chemical itself exhibits no specificity. However, specific antagonists can exploit the difference between the muscarinic and nicotinic receptors. As a result, structure-activity relationships have emerged. All cholinergic agonists appear to need a quaternary ammonium group as well as an atom capable of forming a hydrogen bond through an unshared pair of electrons. The distance between the two may determine whether the agonism is nicotinic or muscarinic. With muscarinic agonists, the distance appears to be about 4.4 Å, whereas for nicotinic agonists, the distance is 5.9 Å.
The nicotinic receptors on ganglia and motor end plates differ, and they are blocked by different drugs. D-Tubocurarine predominantly blocks the neuromuscular junction, whereas hexamethonium acts to block the ganglionic receptors. Methonium compounds were developed to explore the structure-activity relationships of the curare alkaloids. The most potent depolarizing neuromuscular blocking structure contains 10 carbon atoms (i.e., decamethonium); in contrast, the structure containing six carbon atoms (i.e., hexamethonium) is an active ganglionic blocking agent but has little effect at the neuromuscular junction. There is some evidence that ganglionic nicotinic acetylcholine receptors are far more sensitive to anesthetics than receptors in the neuromuscular junction but the clinical significance is unclear.[153] The identification of a new class of acetylcholine-binding proteins released from glial cells suggests another
Acetylcholine receptors on the neuromuscular junctions of mature mammals belong to the superfamily of receptor-gated ion channels, which includes glutamate and glycine. The nicotinic receptors are pentameric membrane proteins, which form nonselective cation channels. There are two α-units (each 40 kd) and one each of the β-, epsilon-, and δ-units ( Fig. 16-19 ). Although different subunits may be expressed developmentally, the α-subunits represent the binding sites for acetylcholine or nicotinic antagonists. At birth, a γ-subunit occupies the position that will be taken by the epsilon-subunit within the first 2 weeks of life. This change in subunits converts the receptor from one with a low conductance and a relatively long duration of opening to a receptor with a high conductance but a brief duration of opening.[155] There are functional differences in acetylcholine receptors during development, but the important drug-binding subunits remain constant.
These five subunits surround each ion channel through which sodium or calcium may enter the cell or potassium may exit. Each ion has its own separate channel, a unique characteristic of the neuromuscular junction. For the channel to open, acetylcholine must occupy a receptor site on each of the two α-subunits. If only one site is occupied and the other is empty, the channel remains closed, and there is no flow of ions and no change in electrical potential. If one site is occupied by acetylcholine and the other site is occupied by an antagonist such as D-tubocurarine, or if both sites are occupied by D-tubocurarine, the channel also remains closed. The ion channel response to acetylcholine is instantaneous and usually lasts only a few milliseconds because acetylcholine is rapidly destroyed by acetylcholinesterase in the synapse. This time course lends a rapidity and flexibility of response of motor end plates to neural stimulation that contributes profoundly to the viability of an
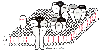 Figure 16-19
Sketch of postjunctional nicotinic acetylcholine receptors
with an acetylcholinesterase (AChoE) molecule nearby. (From Standaert F:
Donuts and holes: Molecules and muscle relaxants. In
Katz R [ed]: Muscle Relaxants: Basic and Clinical Aspects. Orlando, FL, Grune
& Stratton, 1984.)
Figure 16-19
Sketch of postjunctional nicotinic acetylcholine receptors
with an acetylcholinesterase (AChoE) molecule nearby. (From Standaert F:
Donuts and holes: Molecules and muscle relaxants. In
Katz R [ed]: Muscle Relaxants: Basic and Clinical Aspects. Orlando, FL, Grune
& Stratton, 1984.)
In addition to binding of the α-subunit binding site by competing, nonacetylcholine structures, there are two types of channel block: open and closed. With open-channel block, a drug enters a channel opened by acetylcholine but cannot travel all the way through the channel. It impedes ionic flow and prevents depolarization. Because only an open channel may be entered, the intensity of this block depends on how often the channels are open and how active the system is; it is therefore use dependent. Open-channel blockade is driven by the electrical potential difference across the membrane and the charge inherent in the molecular structure. Drugs penetrating an open channel may temporarily bind at some point on the wall of the channel, and the duration of effect therefore partially depends on the identity of the molecule. Closed-channel blockade is harder to study and is less well understood. In this case, a drug may react with the mouth of a closed channel and prevent ion flow. Channel opening is not required, and blockade is therefore not use dependent. Because block does not occur at the acetylcholine receptor site, closed-channel block is not caused by a competitive antagonism of acetylcholine. The classic agents that inhibit cholinesterase may not be completely effective.
In adult skeletal muscle and postganglionic cells, cholinoceptors and their associated ion channels are present only in the immediate synaptic area and are absent from the rest of the cell membrane. The high density (10,000 molecules/µm2 ) of nicotinic acetylcholine receptors at the motor end plate is central to accomplishing successful neuromuscular transmission. Studies have demonstrated that agrin, a nerve-derived extracellular protein in the basal lamina, provides the signal that directs formation of presynaptic terminals[156] [157] and results in a 1000-fold increase in nicotinic receptors in the motor end plate within hours of the arrival of the
Compared with adrenergic receptors, the cholinergic receptors turn over slowly. When a nerve to a muscle is transected, it takes 1 to 3 days to increase the number of cholinergic receptors. In the diaphragm, the number may increase eightfold. These new receptors are no longer confined to the motor end plate, and that change has important clinical ramifications, especially in burn-induced denervation.
In contrast to the ion-gated nicotinic receptors, muscarinic receptors belong to the superfamily of G protein-coupled receptors. Muscarinic receptors have a greater homology to α- and β-adrenergic receptors than to nicotinic receptors. Like the other members of the family of receptors with seven helices (i.e., α2 , β1 , β2 , serotonin, rhodopsin, and opsin), muscarinic receptors use G proteins for signal transduction. Five muscarinic receptors (i.e., M1 through M5 ) exist with the primary structural variability residing in a huge cytoplasmic loop between the fifth and sixth membrane-spanning domains. Although molecular studies have described five forms, of which four are defined pharmacologically (i.e., M1 , M2 , M3 , and M4 ), selective muscarinic drugs are not available. The M2 -cholinergic postjunctional receptor predominates in visceral organs. M2 and M3 receptors have been identified in the airway smooth muscle of many species. In vitro studies reveal that the M3 receptor mediates the contractile and secretory response. However, the excess of M2 receptors could explain the relative ineffectiveness of β-adrenergic agonists in reversing cholinergic bronchoconstriction. [159]
The muscarinic receptors have diverse signal transduction mechanisms. The odd-numbered receptors (i.e., M1 , M3 , and M5 ) work predominantly through the hydrolysis of polyphosphoinositide, whereas the even-numbered receptors work primarily through Gi proteins to regulate adenylate cyclase. [160]
When the M3 muscarinic receptor is activated, Gq activates phospholipase C, which catalyzes the hydrolysis of phosphatidylinositol biphosphate into diacylglycerol and inositol triphosphate. Receptors in the muscarinic series are coupled to second messenger systems, such as cyclic nucleotides or phosphoinositides. These are coupled to ion channels. In other cases, the influx of a cation is the trigger for cellular response. In some cases, however, an influx of calcium ions acts as messengers that react with and open other ion channels. The nature of the response is determined by the specific cation. If calcium or sodium is permitted to flow, the membrane depolarizes; if only potassium is permitted to flow, the membrane hyperpolarizes. In addition to affecting ion channels, the messenger calcium can stimulate various intracellular proteins to alter cell activity. In cardiac atria, activation of muscarinic receptors leads to the efflux of potassium and hyperpolarization of the cell membrane. This efflux slows conduction and slows or stops pacemakers. In glands, an influx of calcium or sodium, or both, activates intracellular events and causes the cells to secrete. Similarly, the influx of these ions into smooth muscle cells causes them to contract.
Muscarinic receptors are found on central and peripheral neurons; a single neuron may have muscarinic receptors with excitatory as well as inhibitory effects. Prejunctional autoreceptors are perhaps not as well studied in the parasympathetic nervous system as in the sympathetic nervous system. Exogenous α2 -agonists may act on prejunctional cholinergic receptors to decrease acetylcholine release. As in the sympathetic system, presynaptic inhibition is of clinical relevance. Presynaptic muscarinic receptors may inhibit the release of acetylcholine from postganglionic parasympathetic neurons; prejunctional nicotinic receptors may increase its release.
Because of the complex coupling, the response of the muscarinic system is sluggish; no response is seen for seconds to minutes after the application of acetylcholine. Similarly, the effect long outlives the presence of the agonist. Even though the transmitter is destroyed rapidly, the train of events it initiates causes the cellular response to continue for many minutes. Muscarinic receptors are desensitized through agonist-dependent phosphorylation in a mechanism similar to that described earlier for β-adrenergic receptors.
|
|
|
|
|
|
|
|
|
|
|
|
|