|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hematologic emergencies in the ICU include abnormalities in coagulation, immunity, and the red blood cell (RBC) mass. These abnormalities can be primary isolated defects, or they can be secondary to multiorgan system failure. The immune system is discussed in the section on infectious disease.
Normal clotting includes an initial phase of platelet hemostatic plug formation and a second stage of fibrin production that occurs by either the intrinsic or extrinsic pathway ( Fig. 76-2 ). For both phases to occur, platelets, coagulation factors, and an intact blood vessel are essential.[300] Neonates have a number of measurable coagulation abnormalities that rarely have clinical manifestations. Term infants and most preterm infants show normal platelet-vessel interaction, but platelet aggregation is transiently impaired. In addition, many coagulation factors are decreased in activity or concentration in the fetus and newborn. Of greatest importance are the vitamin K-dependent factors: factors II, VII, IX, and X. These factors are low at birth and fall to even lower levels during the first week of life unless vitamin K is administered. Factors V and VIII are close to adult levels in all but the most premature infants. Although routine screening tests for coagulation activity are prolonged in infants, the newborn's blood clots more rapidly in vitro because of a deficiency of naturally occurring protease inhibitors, principally antithrombin III.[300]
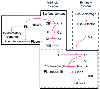 Figure 76-2
Scheme of blood coagulation. The suffix "a" denotes
the activated factor with enzyme activity. HMWK, high-molecular-weight kininogen;
PK, prekallikrein. (Redrawn from Hathaway WE: Hemostasis. In
Rudolph AM [ed]: Pediatrics, 17th ed. East Norwalk, CT, Appleton-Century-Crofts,
1982, p 1110.)
Figure 76-2
Scheme of blood coagulation. The suffix "a" denotes
the activated factor with enzyme activity. HMWK, high-molecular-weight kininogen;
PK, prekallikrein. (Redrawn from Hathaway WE: Hemostasis. In
Rudolph AM [ed]: Pediatrics, 17th ed. East Norwalk, CT, Appleton-Century-Crofts,
1982, p 1110.)
Coagulation disorders are diagnosed on the basis of the history, physical examination, and laboratory data. It is important to elicit any history of easy bleeding, bruising, drug ingestion, associated illnesses, or family history of bleeding. The patient should be closely examined for evidence of petechiae or bruises, bleeding gums, or hepato-splenomegaly. Any sites of bleeding or venipuncture should be observed for fresh clot or oozing. Laboratory evaluation of PTT, PT, thrombin time, platelet count, and bleeding time gives valuable information on the nature of the hemostatic defect.
Hereditary deficiencies of most of the coagulation factors have been described, but classic hemophilia (factor VIII) and Christmas disease (factor IX) account for most (see Chapter 27 ). Since the advent of modern factor replacement therapy, these disorders only rarely cause life-threatening hemorrhage requiring ICU management.[301]
Factor VIII deficiency, or hemophilia A, is transmitted by sex-linked recessive inheritance and occurs in 1 in 10,000 male infants. The severity of the clinical disease is determined by the amount of circulating factor: severe disease is associated with less than 1% of normal levels, moderately severe disease with 1% to 5%, and very mild disease with 5% to 30% of normal levels. The most common sites of bleeding are the joint spaces, but hemorrhage may also occur in the peritoneal cavity, GI tract, muscle, skin, or CNS. Severe trauma or surgery can cause extensive bleeding. The diagnosis is made by demonstrating an elevated PTT
von Willebrand disease occurs nearly as commonly as hemophilia A and is transmitted to both sexes by autosomal dominant inheritance. vWF is a multimeric glycoprotein that is produced by megakaryocytes and endothelial cells. vWF adheres to the subendothelial matrix after vascular injury. It changes conformation and causes platelets to adhere and activate. von Willebrand disease is classified into three types. Type I has a quantitatively reduced amount of normal vWF and accounts for 85% of cases. Type II has qualitatively abnormal vWF, and type III has no vWF. Skin and mucous membrane bleeding, particularly nose-bleeds and menorrhagia, are the most frequent clinical problems. The PTT and bleeding time are prolonged in this disorder, as are some tests of platelet function. Desmopressin can be used to increase plasma vWF in patients with type I and some patients with type II disease. Cryoprecipitate or other plasma derivatives of vWF can be used in patients unresponsive to desmopressin and in those in whom severe bleeding occurs.[303]
Hereditary thrombocytopenia is an uncommon congenital defect. Most cases are associated with familial sex-linked immunodeficiency diseases such as Wiskott-Aldrich syndrome.[304] Platelet transfusions are given to control clinical bleeding, splenectomy often corrects the thrombocytopenia, and successful bone marrow transplantation cures Wiskott-Aldrich syndrome.
A variety of circumstances can impair the production of coagulation factors. The vitamin K-dependent factors are the most commonly affected.[305] These factors are decreased in the presence of liver disease, warfarin therapy, and malabsorption syndromes secondary to either bowel disease or altered bowel flora with long-term antibiotic therapy. In addition, untreated vitamin K deficiency in the neonatal period results in hemorrhagic disease of the newborn. In these disorders, the PT is prolonged, and specific assays demonstrate low levels of factors II, VII, IX, and X. Administration of vitamin K usually reverses these deficiencies unless the synthetic function of the liver is markedly compromised.
Acquired platelet abnormalities include problems of decreased production, increased destruction, and decreased function. Decreased production or hypoproliferative states include marrow diseases such as leukemia and aplastic anemia and, perhaps most commonly, side effects of chemotherapeutic agents. Increased destruction can be immune mediated (i.e., idiopathic thrombocytopenic purpura[306] ) or occur as a result of consumption (i.e., microangiopathic states, HUS, or thrombotic thrombocytopenic purpura[307] ). Finally, platelet dysfunction has been demonstrated in the presence of uremia and chronic polycythemia in patients with cyanotic heart disease.[308]
Treatment of all the acquired thrombocytopenias includes platelet transfusions and, if possible, correction of the underlying disorder. Therapeutic splenectomy has been used to increase platelet survival in some of the severe immune-mediated diseases.
DIC is characterized as a consumptive coagulopathy with decreased platelets and fibrinogen; increased PT, PTT, and thrombin time; and elevated fibrin degradation products. DIC complicates a number of non-specific conditions: sepsis, anaphylaxis, shock, acidosis, massive tissue trauma, sickle cell disease, and certain malignancies, particularly acute myelogenous leukemia (AML) in childhood.[309] Purpura fulminans is a particularly severe type of DIC associated with ecchymosis and thrombosis of the skin, subcutaneous tissue, and distal ends of the extremities. This condition is seen with acute meningococcemia, as well as with other bacterial, viral, and fungal septicemias. Purpura fulminans is associated with high morbidity and mortality; the main morbidity is ischemic loss of the distal ends of the extremities. [310]
Treatment of DIC consists of removing the triggering event whenever and as soon as possible. General support of vascular volume and oxygen transport is essential. When active bleeding is present, transfusion of platelets (0.2 U/kg) and fresh frozen plasma (10 mL/kg) every 4 to 6 hours as a bolus or continuous infusion will replenish platelet and clotting factors, as well as coagulation antagonists such as antithrombin III. Heparin has been advocated by some clinicians; however, it has not been shown to improve outcome and may enhance clinical bleeding. Transfusion with factor concentrates may be helpful when fresh frozen plasma therapy is limited by intravascular hypervolemia. Occasionally, exchange transfusion with fresh whole blood or plasma exchange/plasmapheresis with fresh frozen plasma and platelets may be necessary to treat severe DIC effectively.[311] [312] [313]
|
|
|
|
|
|
|
|
|
|
|
|
|