|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nerve agents are members of a very large group of chemical compounds called organophosphates, known since the 19th century but first examined in detail in the 1930s.[27] More than 50,000 organophosphate compounds have been synthesized, and several are in regular use as insecticides around the world, including parathion, malathion, and fenthion. Although organophosphates were originally developed as insecticides, certain highly toxic members of the class, known as nerve agents, were developed before World War II specifically for military use. As a result of development by Germany and later by Russia, the United States, and Great Britain, at least five nerve agents have been produced. The formulas of the more common nerve agents are shown in Figure 64-3 . The organophosphates tabun (GA), sarin (GB), soman (GD), and cyclosarin (GF) are among the most toxic chemical warfare agents known and together comprise the G-series nerve agents, so named because German scientists first synthesized them, beginning with GA in 1936. GA was followed during World War II by GB and GD. O-ethyl-S-[2(diisopropylamino)ethyl] methylphosphonothioate (coded VX), O-isobutyl S-(2-diethylaminoethyl)methyl phosphothioate (coded VR, also called Russian VX [RVX]), and cyclosarin
 Figure 64-3
Chemical formulas of organophosphate nerve agents: tabun
(GA) (A); sarin (GB) (B);
soman (GD) (C); and VX (D).
(From Baker DJ: Anesthesia in extreme environmental conditions. Part 2.
Chemical and biologic warfare. In Grande CG [ed]:
Textbook of Trauma Anesthesia and Critical Care. Baltimore, Mosby-Year Book, 1993,
p 1331.)
Figure 64-3
Chemical formulas of organophosphate nerve agents: tabun
(GA) (A); sarin (GB) (B);
soman (GD) (C); and VX (D).
(From Baker DJ: Anesthesia in extreme environmental conditions. Part 2.
Chemical and biologic warfare. In Grande CG [ed]:
Textbook of Trauma Anesthesia and Critical Care. Baltimore, Mosby-Year Book, 1993,
p 1331.)
In terms of physical properties, tabun, VX, and VR are persistent, but sarin, soman, and cyclosarin are not. However, soman may be made in a thickened form that is persistent. The relevance of the physicochemical data to the clinical situation is that the nonpersistent agents generally pose a respiratory risk, whereas the persistent agents, which have low vapor pressure, can remain on clothes and other surfaces and thereby present a danger by means of skin contact and absorption. This means that different agents pose specific risks to victims and to medical attendants.
Nerve agents were originally called nerve gases, but they are liquids with volatilities varying between that of petrol and heavy lubricating oil. None of the agents freezes until -40°C. They are pale yellow to colorless, odorless, and soluble in water, in which they undergo slow hydrolysis. However, in the presence of strong alkalis and hypochlorite solution, the hydrolysis is rapid, and this is the basis of the field decontamination of the G nerve agents. Hypochlorite reaction with V nerve agents can itself produce toxic products and is not recommended. Nerve agents can penetrate clothing, leather, and skin. Rubber and synthetic materials such as polyethylene and butyl rubber are more resistant. The physical properties of the nerve agents are shown in Table 64-5 .
The main action of nerve agents is the anticholinesterase effect against acetylcholinesterase and butyrylcholinesterase. Both enzyme systems are familiar to anesthesiologists, who inhibit them on a daily basis using the carbamate anticholinesterase neostigmine to reverse the action of nondepolarizing neuromuscular blocking agents. Organophosphates also inhibit other enzymes, notably neurotoxic esterase. This inhibition causes long-term neurologic effects unrelated to cholinergic changes. The interaction of organophosphates with acetylcholinesterase is complex and is analogous to the interaction of the enzyme with the natural substrate acetylcholine ( Fig. 64-4 ).[3] Inhibition of acetylcholinesterase causes a buildup of acetylcholine at muscarinic and nicotinic synapses of the cholinergic nervous system. There are central and peripheral signs and symptoms that can be directly explained from the classic pharmacologic knowledge of how the cholinergic system operates.
Although the classic effects of organophosphates are cholinergic in nature, there are important effects on other receptor systems, notably γ-aminobutyric acid (GABA) and N-methyl-D-aspartate (NMDA), which give rise to central excitation causing the initial convulsions seen in acute organophosphate poisoning.
The classic signs and symptoms of nerve agent poisoning are shown in Table 64-6 and Table 64-7 . Poisoning is caused by the accumulation of acetylcholine and not by the organophosphate itself. As a result of stimulation of muscarinic synapses, there is miosis, ciliary body spasm causing pain,[28] glandular hypersecretion (including salivary,
| Properties | Tabun (GA) * | Sarin (GB) * | Soman (GD) * | VX * |
|---|---|---|---|---|
| Molecular weight (daltons) | 162.3 | 140.1 | 182.18 | 267.36 |
| Specific gravity at 25°C | 1.073 | 1.0887 | 1.022 | 1.0083 |
| Boiling point (°C) | 246 | 147 | 167 | 300 |
| Melting point (°C) | -49 | -56 | -80 | -20 |
| Vapor pressure and volatality |
|
|
|
|
| 0°C | 0.004 † | 38.0 ‡ | 0.52 † | 4,279.0 ‡ | 0.044 † | 470.9 ‡ | — | — |
| 10°C | 0.013 | 119.5 | 1.07 | 8,494.0 | 0.110 | 1135.5 | — | — |
| 20°C | 0.036 | 319.8 | 2.10 | 16,101.0 | 0.270 | 2692.1 | 0.00044 † | 5.85 ‡ § |
| 25°C | 0.070 | 611.3 | 2.90 | 21,862.0 | 0.400 | 3921.4 | 0.00070 | 10.07 |
| 30°C | 0.094 | 807.4 | 3.93 | 29,138.0 | 0.610 | 5881.4 | — | — |
| 40°C | 0.230 | 1912.4 | 7.10 | 60,959.0 | — | — | — | — |
| 50°C | 0.560 | 4512.0 | 12.30 | 83,548.0 | 2.600 | 23516.0 | — | — |
| From Baker DJ: Anesthesia in extreme environmental conditions. Part 2. Chemical and biologic warfare. In Grande CG (ed): Textbook of Trauma Anesthesia and Critical Care. Baltimore. Mosby-Year Book, 1993, pp 1320–1354. | ||||||||
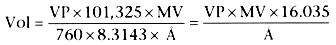
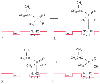 Figure 64-4
Reaction of acetylcholine and sarin with acetylcholinesterase.
(From Baker DJ: Anesthesia in extreme environmental conditions. Part 2.
Chemical and biologic warfare. In Grande CG [ed]:
Textbook of Trauma Anesthesia and Critical Care. Baltimore, Mosby-Year Book, 1993,
p 1341.)
Figure 64-4
Reaction of acetylcholine and sarin with acetylcholinesterase.
(From Baker DJ: Anesthesia in extreme environmental conditions. Part 2.
Chemical and biologic warfare. In Grande CG [ed]:
Textbook of Trauma Anesthesia and Critical Care. Baltimore, Mosby-Year Book, 1993,
p 1341.)
Much of the information about organophosphate actions has been gathered from animal studies and from management of pesticide poisoning of humans. The animal studies do not necessarily represent human reactions, and pesticide studies may not accurately represent the cholinergic syndrome after nerve agent exposure. Although there is some evidence from accidental exposure to sarin and from some human volunteer studies conducted early in the Cold War, the clinical evidence base is smaller than that for pesticide poisoning. During the early stages of the Cold War, several experimental studies were done on human volunteers exposed to nerve agent, and these have been reviewed by Marrs and colleagues.[7] The attacks in Japan in 1994 and 1995 also provided important information concerning the signs and symptoms after sarin release.[25]
The Iran-Iraq War produced first-hand clinical information about the effects and management of nerve agent poisoning, providing a useful platform for modern clinical management.[23] Iranian casualties from nerve agents appeared to have fallen into four broad categories. Those who suffered the greatest exposure died in the field; mask protection by Iranian troops was severely compromised by their having to wear beards for religious reasons. Despite the fact that Iraqi attacks were made against troops having
| Receptor | Target Organ | Symptoms and Signs |
|---|---|---|
| Muscarinic | Iris muscle; ciliary muscle | Miosis; spasm leading to failure of accommodation; headache; pain in the eyes; nausea and vomiting |
|
|
Conjunctival vessels | Vasodilation and hyperemia |
|
|
Nasal glands | Rhinorrhea and hyperemia |
|
|
Bronchial glands | Increased secretion |
|
|
Bronchial muscle | Bronchoconstriction; tightness in the chest; expiratory wheezing; dyspnea |
|
|
Gastrointestinal tract | Anorexia; nausea; vomiting; abdominal cramps; diarrhea; tenesmus; involuntary defecation |
|
|
Sweat glands | Increased activity |
|
|
Salivary glands | Increased activity |
|
|
Lacrimal glands | Lacrimation (not usually marked) |
|
|
Heart | Bradycardia; occasionally tachycardia |
|
|
Bladder | Frequency, involuntary micturition |
| Nicotinic | Skeletal muscle | Weakness; fatigue; fasciculations; cramps; flaccid paralysis (early effects on respiratory muscles may produce dyspnea) |
|
|
Autonomic ganglia | Pallor, occasional elevation of blood pressure |
| Central nervous system muscarinic nicotinic |
|
Anxiety; giddiness; restlessness; headache; withdrawal and depression; memory failure; impaired concentration; slurred speech; depression of respiratory and cardiovascular centers; Cheyne-Stokes respiration |
| From Baker DJ: Anesthesia in extreme environmental conditions. Part 2. Chemical and biologic warfare. In Grande CG (ed): Textbook of Trauma Anesthesia and Critical Care. Baltimore. Mosby-Year Book, 1993, pp 1320–1354. | ||
|
|
|
Symptoms and Signs | |
|---|---|---|---|
| Short-term Ct | AChE Depression (%) | Vapor | Systemic Exposure Only; Eyes Protected |
| <2 | <5% | Incipient miosis (miosis produced at Ct = 2, t = 30 min); slight headache | None |
| 5 | 0–30 | Increased miosis; headache; eye pain, rhinorrhea; conjunctival injection; tightness in chest | Perhaps tightness in chest |
| 5–15 | 10–60 | Eye signs maximal; bronchospasm in some subjects | Symptoms beginning to appear; bronchospasm |
| 15 | 40–60 | Bronchospasm and all the effects described | Wheezing; salivation; eye effects; nausea; vomiting (local sweating and fasciculation in liquid contamination of skin) |
| 40 | 70–90 | Symptoms and signs as for systemic exposure | Weakness; defecation; urination; paralysis; convulsions |
| 100 | 100 | Respiratory failure; death | Respiratory failure; death |
| AChE, acetylcholinesterase; Ct, concentration time. | |||
| From Baker DJ: Anesthesia in extreme environmental conditions. Part 2. Chemical and biologic warfare. In Grande CG (ed): Textbook of Trauma Anesthesia and Critical Care. Baltimore. Mosby-Year Book, 1993, pp 1320–1354. | |||
There is considerable clinical evidence about the effects of organophosphate pesticides from the many thousands of cases that occur in agricultural areas of the world each year.[32] Although these give an overall picture that corresponds to the signs and symptoms described previously, there are probably considerable differences from nerve agents. It is likely that different nerve agents have relatively different effects on the central and nervous systems, and there is evidence for this from rodent studies,[28] although they must be interpreted carefully for human applications.
Critical care management of organophosphate pesticide poisoning has indicated short- and medium-term cardiac changes. After an initial tachycardia (mediated through the anomalous sympathetic nervous system) and a vagally induced bradycardia, there may be ventricular dysrhythmias and prolongation of the QT interval. This has been reported as a poor prognostic sign.[30] [31] [32]
Atropine is a familiar drug in anesthesiology and has long been a mainstay in the management of organophosphate poisoning. Its antagonistic action against acetylcholine at the muscarinic synapses allows control of the muscarinic effects, the most severe of which is bradycardia. Atropine is more beneficial than glycopyrrolate, which has a shorter half-life and does not cross the blood-brain barrier. Atropine has been used for many years in the management of pesticide poisoning, but the relevance of the experience with pesticides to poisoning with nerve agents is not certain.[7] The traditional military response to confirmed or suspected attack with nerve agents is to use
 Figure 64-5
Military automatic intramuscular injection device containing
nerve agent antidotes (Combopen).
Figure 64-5
Military automatic intramuscular injection device containing
nerve agent antidotes (Combopen).
Oximes are compounds capable of reactivating, in some cases, the complex formed by the organophosphate and acetylcholinesterase. Clinically, this means that they can reverse the actions of the organophosphate at the nicotinic receptor and therefore reduce the degree of paralysis.[37] [38] Oximes are widely used in the clinical management of organophosphate pesticide and nerve agent poisoning. Chemically, they are monopyridinium or bispyridinium compounds, which can bind to the complex and cause the nerve agent molecule to separate from the enzyme.
Oximes are a mainstay of the standard military response to nerve agent attack and are widely used in the management of pesticide poisoning. However, their effectiveness depends on the exact nature of the nerve agent involved and on the length of time after the attack before they are given, because chemical changes take place in the nerve agent-enzyme complex known as aging. Aging occurs very rapidly in humans after soman exposure but less so with the other nerve agents. One of the major problems related to oxime research is the selection of a suitable animal model. Originally, monkeys and guinea pigs were thought to be good models for human organophosphate intoxication, and human treatment protocols were developed from such studies. However, there are major differences between aging rates for primates and those for rodents.[39] The rate of formation of aged
Several oximes are available, and recent years have seen the development of new compounds. The most commonly used is pralidoxime (as the chloride or methane sulphonate [mesylate]). Obidoxime is used in some countries. Research has concentrated on the rational use of a range of oximes in terms of the type of oxime used and the dose and timing.[38] [40] HI-6, a Hagedorn oxime, was thought to be able to reactivate the soman-enzyme complex, the most difficult clinical situation to treat. However, there is no evidence that this is true. However, HI-6 may be useful because of other pharmacologic properties and may be advantageous in the treatment of cyclosarin poisoning and other cases in which obidoxime was previously used.
For the average anesthesiologist treating a patient with the symptoms of nerve agent poisoning, a civil setting is the most likely. In this situation, exposure to sarin is the most probable situation, and treatment schedules should logically be based on this premise. Military anesthesiologists may face other hazards but are provided with specialized detection equipment and treatment modalities based on a threat assessment. In the civil context, all nerve agent casualties should receive pralidoxime mesylate initially in addition to atropine and ventilatory life support.
Some authorities believe that HI-6 should replace pralidoxime when regular supplies are available. Obidoxime was thought to be particularly effective against tabun poisoning, but there is no specific evidence to support this belief.
Oxime therapy should be given simultaneously with atropine.[41] Slow intravenous injection of pralidoxime is recommended to prevent laryngospasm, muscle rigidity, and hypertension. Pralidoxime (15 to 30 mg/kg) is given intramuscularly or intravenously over 20 minutes for adults and children. This dose may be repeated after 4 hours (or 1 hour if paralysis is worsening). The target therapeutic blood concentration should be 4 µg/mL. However, studies by Worek and his group[38] showed that the full therapeutic effects of pralidoxime might be achieved at higher concentrations. The same group showed that there was a possibility of repeat inhibition of acetylcholinesterase after pralidoxime treatment in human erthrocytes, but the relevance to the whole body is unclear. Sarin is broken down rapidly in the blood by hydrolysis. Oxime treatment in hospital should continue for as long as atropine is required.
The central actions of organophosphates give rise to spike discharges and, in extreme cases, convulsions. Benzodiazepines have long been used to counter this action. Given the known action of organophosphates at the GABA receptor and the antagonistic action of benzodiazepines at this site, the action may be noncholinergic. [42]
The problem of aging of the organophosphate-acetylcholinesterase complex, particularly with soman, gave rise to a novel approach to prophylaxis against nerve agent poisoning in the military context.[8] Although pyridostigmine pretreatment is unlikely to be used in mass treatment of civilian populations, there are important consequences for anesthesia, and all civilian anesthesiologists should be aware of the side effects.
Pyridostigmine is a dimethyl carbamate compound with a quaternary nitrogen atom. As a result, it does not penetrate the blood-brain barrier to any extent. In common with other carbamates such as neostigmine and physostigmine, pyridostigmine is an anticholinesterase and has essentially the same action as organophosphates. However, in the case of carbamates, the complex formed with the enzyme is readily reversible. The normal treatment dose of pyridostigmine is 30 mg every 8 hours. There is no plasma protein binding and no drug interactions involving competition for binding sites. Between 79% and 90% of the absorbed dose is excreted unchanged in the urine. The reversibility of the complex formed with acetylcholinesterase is indicated in that at the usual pretreatment level, the enzyme level falls to 10% of normal within 12 hours after the last dose. At steady state, only 40% of the available acetylcholinesterase is complexed. Given the considerable safety margin that exists in the level of enzyme at cholinergic synapses, treatment with pyridostigmine does not produce any more than mild parasympathetic signs.[43]
Pyridostigmine has a protective action against organophosphates because the carbamylate-complexed acetylcholinesterase resists attack after subsequent exposure to an organophosphate. If a person taking pyridostigmine is exposed later to a potentially lethal dose of organophosphate, a reserve of enzyme is effectively autotransfused into the patient during the subsequent period. This process does not require the presence of an oxime. The organophosphate is rapidly hydrolyzed in the bloodstream after exposure and has no further action on the released acetylcholinesterase. The amount of enzyme released from the carbamylate complex is sufficient to restore neuromuscular transmission because of the safety margin that exists at the synapse. However, in severe cases of organophosphate poisoning, life-support measures are necessary to bridge the gap between the organophosphate's attack on the synapse and restoration of the acetylcholinesterase level through the autotransfusion.
The anticholinergic actions of pyridostigmine have considerable bearing on the practice of anesthesia. Butyrylcholinesterase, a determinant of the metabolism of succinyl choline, is also inhibited, and prolonged action of this drug may be expected in patients taking pyridostigmine.[44] Carbamate neostigmine is usually given at the end of an operation to reverse the action of nondepolarizing, neuromuscular-blocking drugs. Pretreatment with a carbamate of a patient who is not subsequently exposed to an organophosphate may be expected to produce some resistance to the action of the neuromuscular blocker. Experimental studies[45] indicate that pyridostigmine does not alter the overall characteristics of neuromuscular block in an isolated human forearm, and the clinical effects may be minimal, particularly because more central muscles of respiration such as the diaphragm
|
|
|
|
|
|
|
|
|
|
|
|
|