ATHEROGENESIS
Atherosclerosis is a progressive disease process that compromises
the arterial blood supply to any or all of the vital organs or the extremities.
Risk factors include hypercholesterolemia, diabetes, obesity, hypertension, male
gender, cigarette smoking, and family history. Because atherosclerosis usually has
no early warning signs, prevention or risk modification is difficult until the patient
has progressed to an advanced stage of disease. The lesions of atherosclerosis occur
primarily in large and medium sized arteries and may be present throughout one's
lifetime. The progression of atherosclerosis occurs in three stages.[1]
The initial lesion of atherosclerosis, the fatty streak, starts in early childhood
and forms beneath the normal endothelium. These lesions consist of lipid-laden macrophages
(i.e., foam cells), smooth muscle cells, and elastic and collagen fibers. The fatty
streak progresses to a fibrous plaque consisting of degenerated foam cells covered
by a layer of proliferated smooth muscle cells. Unlike the fatty streak, these lesions
may compromise blood flow through a vascular bed and cause ischemia or thrombosis
to the vital organs or extremities, resulting in infarction. The advanced lesion
of atherosclerosis represents a progression of the fibrous plaque, with an expanded
lipid-rich core, accumulation of calcium, disruption of endothelial integrity, and
platelet thrombus and hemorrhage into the lesion.
The exact mechanism of atherogenesis and the explanation for the
anatomic distribution of lesions are unknown. Several hypothesizes have been offered.
The lipid hypothesis, based on substantial experimental
and clinical associations between hyperlipidemia and atherosclerosis, was dominant
until the 1970s.[2]
The response
to injury hypothesis, which was proposed by Virchow[3]
nearly 150 years ago, has recently come back into favor.[4]
[5]
The hypothesis is that endothelial injury is
caused by a variety of chemical and physical factors, leading to endothelial denudation
and exposure of the subendothelium. Platelet adherence and degranulation stimulate
arterial smooth muscle migration from the media into the intima, a phenomenon that
promotes intimal smooth muscle proliferation. The latest version of this hypothesis
suggests that uninjured but "dysfunctional" endothelium initiates atherogenesis.
[6]
The monoclonal hypothesis
of atherogenesis, proposed by Benditt and Benditt[7]
in 1973, suggests that each atherosclerotic lesion is derived from a single smooth
muscle cell that proliferates around a focus. This hypothesis draws an analogy to
malignancy, in which a cell proliferates uncontrollably in response to a mutagen
or possibly to a virus to produce a lesion. The clonal senescence
hypothesis proposed by Martin and Sprague[8]
also focuses on smooth muscle cell proliferation that is controlled by local feedback
from inhibitory hormones. In aging vessels, a decrease in inhibitory feedback stimulates
growth of these cells and formation of the lesion.
Over the past decade, accumulating experimental and clinical evidence
indicates that inflammation in the artery wall plays a fundamental role in the initiation
and progression of atherosclerosis.[6]
[9]
As a result of this new understanding, inflammation has become a therapeutic target
in the prevention and treatment of atherosclerosis and its complications. Established
pharmacologic strategies for preventing recurrent myocardial infarction (MI) and
death may have unexpected anti-inflammatory effects. For example, inhibitors of
hydroxymethylglutaryl coenzyme A (i.e., statins) reduce low-density-lipoprotein cholesterol
levels but also have anti-inflammatory actions such as reduction in leukocyte adhesion
and macrophage activation.[10]
Based on the substantial
evidence supporting a role for inflammation in the pathogenesis of atherosclerosis,
serum markers of inflammation are being used as markers of atherosclerosis risk.
Elevated plasma levels of C-reactive protein (CRP), an acute-phase reactant produced
in response to inflammatory cytokines (primarily interleukin-6), are being used in
cardiovascular risk stratification.[11]
[12]
CRP levels have been correlated with risk of death and MI[13]
and with the development of peripheral vascular disease.[14]
Aspirin's ability to reduce MI probably reflects its antiplatelet and anti-inflammatory
actions (i.e., reduces CRP levels).[15]
The anatomic distribution of atherosclerotic lesions is shown
in Figure 52-1
. The most
common sites are the coronary arteries, the carotid bifurcation, the abdominal aorta,
and the iliac and femoral arteries.[1]
It is known
that atherosclerotic lesions tend to develop near arterial branch points and along
the outer surfaces of arterial curves. Their development may be related to increased
shear stress and injury to the endothelial surface.[16]
There is a well established and strong associated between elevated cholesterol levels
and atherosclerosis.[17]
Statins are the lipid-modifying
agents of choice for the treatment and prevention of atherosclerosis. The efficacy
of statins at reducing cholesterol levels and cardiovascular events is well established
in large outcome-based clinical trials.[18]
[19]
[20]
When cholesterol level is lowered to less
than
150 mg/dL, lipids are mobilized from atherosclerotic lesions.[21]
Estrogens protect against atherosclerosis in part by increasing high-density lipoproteins
and decreasing low-density lipoproteins.[22]
Some
evidence supports the role of antioxidants in preventing atherosclerotic lesions.
[23]
Vitamins C and E, carotenoids, and folic acid
may be beneficial in this regard.[24]
Aspirin,
by means of its anti-inflammatory and antiplatelet effects, is effective in preventing
stroke and MI. A review of risk and prevention of arterial aging has been published
[24]
and supplemental information is also available
on the Internet.[25]
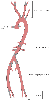 Figure 52-1
Distribution of atherosclerotic lesions. Plaques occur
at the origins of large vessels and at bifurcations. Distribution and relative severity
depend on risk factors. (From Zwolak RM, Cronenwett JL: Pathophysiology
of vascular disease. In Yeager MP, Glass DD [eds]:
Anesthesiology and Vascular Surgery. East Norwalk, CT, Appleton & Lange, 1990,
pp 3–29.)
Figure 52-1
Distribution of atherosclerotic lesions. Plaques occur
at the origins of large vessels and at bifurcations. Distribution and relative severity
depend on risk factors. (From Zwolak RM, Cronenwett JL: Pathophysiology
of vascular disease. In Yeager MP, Glass DD [eds]:
Anesthesiology and Vascular Surgery. East Norwalk, CT, Appleton & Lange, 1990,
pp 3–29.)